![]() –ü—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ–Ĺ–Ĺ–į—Ź —Ā—ā—Ä—É–ļ—ā—É—Ä–į —ć—ā–ł–Ľ–Ķ–Ĺ–į.
–ü—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ–Ĺ–Ĺ–į—Ź —Ā—ā—Ä—É–ļ—ā—É—Ä–į —ć—ā–ł–Ľ–Ķ–Ĺ–į.
–ź–Ľ–ļ–ĶŐĀ–Ĺ—č (—ć—ā–ł–Ľ–ĶŐĀ–Ĺ–ĺ–≤—č–Ķ —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺŐĀ–ī—č) ‚ÄĒ –į—Ü–ł–ļ–Ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –Ĺ–Ķ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ć–Ĺ—č–Ķ —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī—č, —Ā–ĺ–ī–Ķ—Ä–∂–į—Č–ł–Ķ –ĺ–ī–Ĺ—É –ī–≤–ĺ–Ļ–Ĺ—É—é —Ā–≤—Ź–∑—Ć –ľ–Ķ–∂–ī—É –į—ā–ĺ–ľ–į–ľ–ł —É–≥–Ľ–Ķ—Ä–ĺ–ī–į, –ĺ–Ī—Ä–į–∑—É—é—Č–ł–Ķ –≥–ĺ–ľ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ļ —Ä—Ź–ī —Ā –ĺ–Ī—Č–Ķ–Ļ —Ą–ĺ—Ä–ľ—É–Ľ–ĺ–Ļ CnH2n.
–ü—Ä–ĺ—Ā—ā–Ķ–Ļ—ą–ł–ľ –į–Ľ–ļ–Ķ–Ĺ–ĺ–ľ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —ć—ā–ł–Ľ–Ķ–Ĺ (C2H4). –ź—ā–ĺ–ľ—č —É–≥–Ľ–Ķ—Ä–ĺ–ī–į –Ņ—Ä–ł –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –Ĺ–į—Ö–ĺ–ī—Ź—ā—Ā—Ź –≤ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–ł sp2-–≥–ł–Ī—Ä–ł–ī–ł–∑–į—Ü–ł–ł –ł –ł–ľ–Ķ—é—ā –≤–į–Ľ–Ķ–Ĺ—ā–Ĺ—č–Ļ —É–≥–ĺ–Ľ 120¬į.
–ü–ĺ –Ĺ–ĺ–ľ–Ķ–Ĺ–ļ–Ľ–į—ā—É—Ä–Ķ –ė–ģ–ü–ź–ö –ī–Ľ—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ —Ā –Ľ–ł–Ĺ–Ķ–Ļ–Ĺ–ĺ–Ļ (–Ĺ–Ķ—Ä–į–∑–≤–Ķ—ā–≤–Ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ) —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ—Ć—é –Ĺ–į–∑–≤–į–Ĺ–ł–Ķ –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ĺ –ĺ—ā –Ĺ–į–∑–≤–į–Ĺ–ł—Ź –į–Ľ–ļ–į–Ĺ–į —Ā —ā–Ķ–ľ –∂–Ķ —á–ł—Ā–Ľ–ĺ–ľ –į—ā–ĺ–ľ–ĺ–≤ —É–≥–Ľ–Ķ—Ä–ĺ–ī–į –Ņ—É—ā–Ķ–ľ –∑–į–ľ–Ķ–Ĺ–ĺ–Ļ —Ā—É—Ą—Ą–ł–ļ—Ā–į ¬ę-–į–ŬĽ –Ĺ–į ¬ę-–Ķ–ŬĽ —Ā —É–ļ–į–∑–į–Ĺ–ł–Ķ–ľ –Ņ—Ä–ł –Ĺ–Ķ–ľ –Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł—Ź –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –≤ —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł[1]. –Ě–į–Ņ—Ä–ł–ľ–Ķ—Ä: CH2=CH-CH2-CH3 ‚Äď –Ī—É—ā–Ķ–Ĺ-1.‚ě§
–ď–ĺ–ľ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ļ —Ä—Ź–ī –ł –ł–∑–ĺ–ľ–Ķ—Ä–ł—Ź
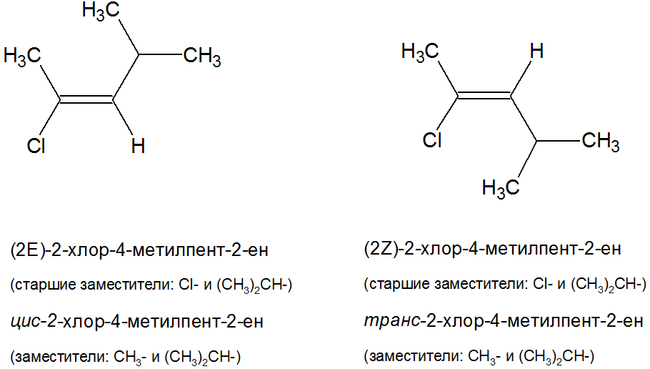
–ź–Ľ–ļ–Ķ–Ĺ—č, —á–ł—Ā–Ľ–ĺ –į—ā–ĺ–ľ–ĺ–≤ —É–≥–Ľ–Ķ—Ä–ĺ–ī–į –≤ –ļ–ĺ—ā–ĺ—Ä—č—Ö –Ī–ĺ–Ľ—Ć—ą–Ķ –ī–≤—É—Ö, (—ā–ĺ –Ķ—Ā—ā—Ć –ļ—Ä–ĺ–ľ–Ķ —ć—ā–ł–Ľ–Ķ–Ĺ–į) –ł–ľ–Ķ—é—ā —Ā–≤–ĺ–ł –ł–∑–ĺ–ľ–Ķ—Ä—č. –Ē–Ľ—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ—č –ł–∑–ĺ–ľ–Ķ—Ä–ł—Ź —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–≥–ĺ —Ā–ļ–Ķ–Ľ–Ķ—ā–į, –Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł—Ź –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł, –ľ–Ķ–∂–ļ–Ľ–į—Ā—Ā–ĺ–≤–į—Ź –ł –≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–į—Ź. –Ě–į–Ņ—Ä–ł–ľ–Ķ—Ä, –Ķ–ī–ł–Ĺ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ –ł–∑–ĺ–ľ–Ķ—Ä–ĺ–ľ –Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ü–ł–ļ–Ľ–ĺ–Ņ—Ä–ĺ–Ņ–į–Ĺ (C3H6) –Ņ–ĺ –ľ–Ķ–∂–ļ–Ľ–į—Ā—Ā–ĺ–≤–ĺ–Ļ –ł–∑–ĺ–ľ–Ķ—Ä–ł–ł. –Ě–į—á–ł–Ĺ–į—Ź —Ā –Ī—É—ā–ł–Ľ–Ķ–Ĺ–į, —Ā—É—Č–Ķ—Ā—ā–≤—É—é—ā –ł–∑–ĺ–ľ–Ķ—Ä—č –Ņ–ĺ –Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł—é –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł (–Ī—É—ā–Ķ–Ĺ-1 –ł –Ī—É—ā–Ķ–Ĺ-2), –Ņ–ĺ —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–ľ—É —Ā–ļ–Ķ–Ľ–Ķ—ā—É (–ł–∑–ĺ–Ī—É—ā–ł–Ľ–Ķ–Ĺ –ł–Ľ–ł –ľ–Ķ—ā–ł–Ľ–Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ) –ł –≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ł–Ķ –ł–∑–ĺ–ľ–Ķ—Ä—č (—Ü–ł—Ā-–Ī—É—ā–Ķ–Ĺ-2 –ł —ā—Ä–į–Ĺ—Ā-–Ī—É—ā–Ķ–Ĺ-2). –° —Ä–ĺ—Ā—ā–ĺ–ľ —á–ł—Ā–Ľ–į –į—ā–ĺ–ľ–ĺ–≤ —É–≥–Ľ–Ķ—Ä–ĺ–ī–į –≤ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–Ķ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ –ł–∑–ĺ–ľ–Ķ—Ä–ĺ–≤ –≤–ĺ–∑—Ä–į—Ā—ā–į–Ķ—ā –≤ –≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł–ł.

–ď–ĺ–ľ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ļ —Ä—Ź–ī –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤:
–ź–Ľ–ļ–Ķ–Ĺ—č –ľ–ĺ–≥—É—ā —Ā—É—Č–Ķ—Ā—ā–≤–ĺ–≤–į—ā—Ć –≤ –≤–ł–ī–Ķ –Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –ł–Ľ–ł –≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ł—Ö –ł–∑–ĺ–ľ–Ķ—Ä–ĺ–≤.
–†–į–∑–Ľ–ł—á–į—é—ā:
- —Ü–ł—Ā- –ł–∑–ĺ–ľ–Ķ—Ä—č: –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–ł —Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ—č –Ņ–ĺ –ĺ–ī–Ĺ—É —Ā—ā–ĺ—Ä–ĺ–Ĺ—É –ĺ—ā –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł;
- —ā—Ä–į–Ĺ—Ā- –ł–∑–ĺ–ľ–Ķ—Ä—č: –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–ł —Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ—č –Ņ–ĺ —Ä–į–∑–Ĺ—č–Ķ —Ā—ā–ĺ—Ä–ĺ–Ĺ—č –ĺ—ā –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł.
IUPAC —Ä–Ķ–ļ–ĺ–ľ–Ķ–Ĺ–ī—É–Ķ—ā –Ĺ–į–∑—č–≤–į—ā—Ć –≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ł–Ķ –ł–∑–ĺ–ľ–Ķ—Ä—č –Ņ–ĺ —Ā–Ľ–Ķ–ī—É—é—Č–Ķ–Ļ –Ĺ–ĺ–ľ–Ķ–Ĺ–ļ–Ľ–į—ā—É—Ä–Ķ:
- Z- –ł–∑–ĺ–ľ–Ķ—Ä—č: —Ā—ā–į—Ä—ą–ł–Ķ –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–ł —É —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ—č—Ö –į—ā–ĺ–ľ–ĺ–≤ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –Ĺ–į—Ö–ĺ–ī—Ź—ā—Ā—Ź –Ņ–ĺ –ĺ–ī–Ĺ—É —Ā—ā–ĺ—Ä–ĺ–Ĺ—É –ĺ—ā–Ĺ–ĺ—Ā–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł;
- E- –ł–∑–ĺ–ľ–Ķ—Ä—č: —Ā—ā–į—Ä—ą–ł–Ķ –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–ł —É —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ—č—Ö –į—ā–ĺ–ľ–ĺ–≤ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –Ĺ–į—Ö–ĺ–ī—Ź—ā—Ā—Ź –Ņ–ĺ —Ä–į–∑–Ĺ—č–Ķ —Ā—ā–ĺ—Ä–ĺ–Ĺ—č –ĺ—ā–Ĺ–ĺ—Ā–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł.
–Ě–ĺ–ľ–Ķ–Ĺ–ļ–Ľ–į—ā—É—Ä–į
–°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –Ĺ–ĺ–ľ–Ķ–Ĺ–ļ–Ľ–į—ā—É—Ä–Ķ IUPAC, –Ĺ–į–∑–≤–į–Ĺ–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ĺ–Ī—Ä–į–∑—É—é—ā –ī–ĺ–Ī–į–≤–Ľ–Ķ–Ĺ–ł–Ķ–ľ —Ā—É—Ą—Ą–ł–ļ—Ā–į ¬ę-–Ķ–ŬĽ –ļ –ĺ—Ā–Ĺ–ĺ–≤–Ķ –Ĺ–į–∑–≤–į–Ĺ–ł—Ź, –ĺ—ā—Ä–į–∂–į—é—Č–Ķ–Ļ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ –į—ā–ĺ–ľ–ĺ–≤ —É–≥–Ľ–Ķ—Ä–ĺ–ī–į –≤ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł. –ü–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –≤ —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł –ĺ–Ī–ĺ–∑–Ĺ–į—á–į–Ķ—ā—Ā—Ź –į—Ä–į–Ī—Ā–ļ–ł–ľ–ł —Ü–ł—Ą—Ä–į–ľ–ł –Ņ–ĺ—Ā–Ľ–Ķ –ĺ—Ā–Ĺ–ĺ–≤—č –ł –Ņ–Ķ—Ä–Ķ–ī —Ā—É—Ą—Ą–ł–ļ—Ā–ĺ–ľ ¬ę-–Ķ–ŬĽ –≤ –į–Ĺ–≥–Ľ–ĺ—Ź–∑—č—á–Ĺ—č—Ö –Ĺ–į–∑–≤–į–Ĺ–ł—Ź—Ö –ł –Ņ–ĺ—Ā–Ľ–Ķ —Ā—É—Ą—Ą–ł–ļ—Ā–į ¬ę-–Ķ–ŬĽ ‚Äď –≤ —Ä—É—Ā—Ā–ļ–ĺ—Ź–∑—č—á–Ĺ—č—Ö. –Ě–į–∑–≤–į–Ĺ–ł—Ź —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī–Ĺ—č—Ö –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–Ķ–Ļ –Ņ—Ä–ł –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł —É–ļ–į–∑—č–≤–į—é—ā—Ā—Ź –≤ –Ņ—Ä–ł—Ā—ā–į–≤–ĺ—á–Ĺ–ĺ–Ļ —á–į—Ā—ā–ł –Ĺ–į–∑–≤–į–Ĺ–ł—Ź. –Ě–į–Ņ—Ä–ł–ľ–Ķ—Ä, CH2=C(CH3)-CH(CH3)-CH3 ¬ę2,3-–ī–ł–ľ–Ķ—ā–ł–Ľ–Ī—É—ā–Ķ–Ĺ-1¬Ľ .
–£–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī–Ĺ—č–Ķ —Ä–į–ī–ł–ļ–į–Ľ—č (–∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–ł), –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–Ĺ—č–Ķ –ĺ—ā –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤, –ł–ľ–Ķ—é—ā –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ļ —Ā—É—Ą—Ą–ł–ļ—Ā ¬ę-–Ķ–Ĺ–łŐĀ–Ľ¬Ľ, (–Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä,¬ę–Ī—É—ā-3-–Ķ–Ĺ-1-–ł–Ľ¬Ľ –ī–Ľ—Ź CH2=CH‚ÄďCH2‚ÄďCH2‚ÄĒ). –Ē–ĺ–Ņ—É—Ā—ā–ł–ľ—č–ľ–ł —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –ī–≤–į —ā—Ä–ł–≤–ł–į–Ľ—Ć–Ĺ—č—Ö –Ĺ–į–∑–≤–į–Ĺ–ł—Ź: CH2=CH‚ÄĒ ¬ę–≤–ł–Ĺ–łŐĀ–Ľ¬Ľ –ł CH2=CH‚ÄďCH2‚ÄĒ ¬ę–į–Ľ–Ľ–łŐĀ–Ľ¬Ľ.
–≠–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–Ĺ–ĺ–Ķ —Ā—ā—Ä–ĺ–Ķ–Ĺ–ł–Ķ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł
–í —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ł–ł —Ā —ā–Ķ–ĺ—Ä–ł–Ķ–Ļ –≥–ł–Ī—Ä–ł–ī–ł–∑–į—Ü–ł–ł –ī–≤–ĺ–Ļ–Ĺ–į—Ź —Ā–≤—Ź–∑—Ć –ĺ–Ī—Ä–į–∑—É–Ķ—ā—Ā—Ź –∑–į —Ā—á—Ď—ā –Ņ–Ķ—Ä–Ķ–ļ—Ä—č–≤–į–Ĺ–ł—Ź –≤–ī–ĺ–Ľ—Ć –Ľ–ł–Ĺ–ł–ł —Ā–≤—Ź–∑–ł –°-–° sp2-–≥–ł–Ī—Ä–ł–ī–Ĺ—č—Ö –ĺ—Ä–Ī–ł—ā–į–Ľ–Ķ–Ļ –į—ā–ĺ–ľ–ĺ–≤ —É–≥–Ľ–Ķ—Ä–ĺ–ī–į (ŌÉ-—Ā–≤—Ź–∑—Ć) –ł –Ī–ĺ–ļ–ĺ–≤–ĺ–≥–ĺ –Ņ–Ķ—Ä–Ķ–ļ—Ä—č–≤–į–Ĺ–ł—Ź —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ—č—Ö p-–ĺ—Ä–Ī–ł—ā–į–Ľ–Ķ–Ļ (ŌÄ-—Ā–≤—Ź–∑—Ć).
 –°—Ö–Ķ–ľ–į –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź —Ā–≤—Ź–∑–Ķ–Ļ –≤ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–Ķ —ć—ā–ł–Ľ–Ķ–Ĺ–į
–°—Ö–Ķ–ľ–į –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź —Ā–≤—Ź–∑–Ķ–Ļ –≤ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–Ķ —ć—ā–ł–Ľ–Ķ–Ĺ–į
–í —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–ł sp2-–≥–ł–Ī—Ä–ł–ī–ł–∑–į—Ü–ł–ł —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–Ĺ–ĺ–Ķ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ –į—ā–ĺ–ľ–į —É–≥–Ľ–Ķ—Ä–ĺ–ī–į –ľ–ĺ–∂–Ĺ–ĺ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–ł—ā—Ć —Ā–Ľ–Ķ–ī—É—é—Č–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ:

–í—Ā–Ķ –į—ā–ĺ–ľ—č —ć—ā–ł–Ľ–Ķ–Ĺ–į –Ľ–Ķ–∂–į—ā –≤ –ĺ–ī–Ĺ–ĺ–Ļ –Ņ–Ľ–ĺ—Ā–ļ–ĺ—Ā—ā–ł, –į –≤–Ķ–Ľ–ł—á–ł–Ĺ–į –≤–į–Ľ–Ķ–Ĺ—ā–Ĺ–ĺ–≥–ĺ —É–≥–Ľ–į —Ā–≤—Ź–∑–ł C-H –Ņ—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł —Ä–į–≤–Ĺ–į 120 ¬į. –¶–Ķ–Ĺ—ā—Ä—č —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ—č—Ö –į—ā–ĺ–ľ–ĺ–≤ –≤ —ć—ā–ł–Ľ–Ķ–Ĺ–Ķ –Ĺ–į—Ö–ĺ–ī—Ź—ā—Ā—Ź –Ĺ–į —Ä–į—Ā—Ā—ā–ĺ—Ź–Ĺ–ł–ł 0,134 –Ĺ–ľ, —ā–ĺ –Ķ—Ā—ā—Ć –ī–Ľ–ł–Ĺ–į –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ĺ –ļ–ĺ—Ä–ĺ—á–Ķ, —á–Ķ–ľ –°-–°.
–°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ —ā–Ķ–ĺ—Ä–ł–ł –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ—č—Ö –ĺ—Ä–Ī–ł—ā–į–Ľ–Ķ–Ļ –Ľ–ł–Ĺ–Ķ–Ļ–Ĺ–į—Ź –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł—Ź –ī–≤—É—Ö –į—ā–ĺ–ľ–Ĺ—č—Ö 2p-–ĺ—Ä–Ī–ł—ā–į–Ľ–Ķ–Ļ —É–≥–Ľ–Ķ—Ä–ĺ–ī–į —Ą–ĺ—Ä–ľ–ł—Ä—É–Ķ—ā –ī–≤–Ķ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ—č–Ķ ŌÄ-–ĺ—Ä–Ī–ł—ā–į–Ľ–ł —ć—ā–ł–Ľ–Ķ–Ĺ–į[2]:
 –§–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ ŌÄ-–ĺ—Ä–Ī–ł—ā–į–Ľ–Ķ–Ļ —ć—ā–ł–Ľ–Ķ–Ĺ–į
–§–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ ŌÄ-–ĺ—Ä–Ī–ł—ā–į–Ľ–Ķ–Ļ —ć—ā–ł–Ľ–Ķ–Ĺ–į
–ü–Ķ—Ä–≤—č–Ļ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ –ł–ĺ–Ĺ–ł–∑–į—Ü–ł–ł —ć—ā–ł–Ľ–Ķ–Ĺ–į —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ķ—ā 10,51 —ć–í[3], —á—ā–ĺ –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź–Ķ—ā —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ—É –ĺ—ā–Ĺ–ĺ—Ā–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –Ľ–Ķ–≥–ļ–ĺ —É—Ö–ĺ–ī–ł—ā—Ć (—ć–Ľ–Ķ–ļ—ā—Ä–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–Ķ –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ) —Ā –≤—č—Ā—ą–Ķ–Ļ –∑–į–Ĺ—Ź—ā–ĺ–Ļ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–Ļ –ĺ—Ä–Ī–ł—ā–į–Ľ–ł (–í–ó–ú–ě).
–í —ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź, –Ĺ–ł–∑—ą–į—Ź —Ā–≤—Ź–∑—č–≤–į—é—Č–į—Ź –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–į—Ź –ĺ—Ä–Ī–ł—ā–į–Ľ—Ć (–Ě–°–ú–ě) —ć—ā–ł–Ľ–Ķ–Ĺ–į –ł–ľ–Ķ–Ķ—ā –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –Ĺ–ł–∑–ļ—É—é —ć–Ĺ–Ķ—Ä–≥–ł—é: ‚ąí1,6‚ÄĒ1,8 —ć–í, —á—ā–ĺ –ĺ–Ī—ä—Ź—Ā–Ĺ—Ź–Ķ—ā –ĺ—ā–Ĺ–ĺ—Ā–ł—ā–Ķ–Ľ—Ć–Ĺ—É—é –Ľ—Ď–≥–ļ–ĺ—Ā—ā—Ć –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź —ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–į —Ā –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –į–Ĺ–ł–ĺ–Ĺ–į[3] (–Ĺ—É–ļ–Ľ–Ķ–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–Ķ –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ).
–Ē–ĺ–Ī–į–≤–Ľ–Ķ–Ĺ–ł–Ķ –ľ–Ķ—ā–ł–Ľ—Ć–Ĺ–ĺ–≥–ĺ –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ—Ź —Ā–Ĺ–ł–∂–į–Ķ—ā –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ –ł–ĺ–Ĺ–ł–∑–į—Ü–ł–ł ŌÄ-—ć–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–ĺ–≤ –Ņ—Ä–ł–ľ–Ķ—Ä–Ĺ–ĺ –Ĺ–į 0,6‚ÄĒ0,8 —ć–í –ł –Ņ–ĺ–≤—č—ą–į–Ķ—ā —ć–Ĺ–Ķ—Ä–≥–ł—é –Ě–°–ú–ě –Ĺ–į 0,2 —ć–í, –į –í–ó–ú–ě –Ĺ–į 0,7 —ć–í[3].
–ė—Ā—ā–ĺ—Ä–ł—Ź –ĺ—ā–ļ—Ä—č—ā–ł—Ź
–í–Ņ–Ķ—Ä–≤—č–Ķ —ć—ā–ł–Ľ–Ķ–Ĺ –Ī—č–Ľ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ –≤ 1669 –≥–ĺ–ī—É –Ĺ–Ķ–ľ–Ķ—Ü–ļ–ł–ľ —Ö–ł–ľ–ł–ļ–ĺ–ľ –ł –≤—Ä–į—á–ĺ–ľ –ė. –ė. –Ď–Ķ—Ö–Ķ—Ä–ĺ–ľ –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ —Ā–Ķ—Ä–Ĺ–ĺ–Ļ –ļ–ł—Ā–Ľ–ĺ—ā—č –Ĺ–į —ć—ā–ł–Ľ–ĺ–≤—č–Ļ —Ā–Ņ–ł—Ä—ā. –£—á—Ď–Ĺ—č–Ļ —É—Ā—ā–į–Ĺ–ĺ–≤–ł–Ľ, —á—ā–ĺ –Ķ–≥–ĺ ¬ę–≤–ĺ–∑–ī—É—Ö¬Ľ –Ī–ĺ–Ľ–Ķ–Ķ —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł –į–ļ—ā–ł–≤–Ķ–Ĺ, —á–Ķ–ľ –ľ–Ķ—ā–į–Ĺ, –ĺ–ī–Ĺ–į–ļ–ĺ –ł–ī–Ķ–Ĺ—ā–ł—Ą–ł—Ü–ł—Ä–ĺ–≤–į—ā—Ć –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–Ĺ—č–Ļ –≥–į–∑ –ĺ–Ĺ –Ĺ–Ķ —Ā–ľ–ĺ–≥ –ł –Ĺ–į–∑–≤–į–Ĺ–ł—Ź –Ķ–ľ—É –Ĺ–Ķ –Ņ—Ä–ł—Ā–≤–ĺ–ł–Ľ[4].
–í—ā–ĺ—Ä–ł—á–Ĺ–ĺ –ł —ā–Ķ–ľ –∂–Ķ —Ā–Ņ–ĺ—Ā–ĺ–Ī–ĺ–ľ ¬ę–≤–ĺ–∑–ī—É—Ö –Ď–Ķ—Ö–Ķ—Ä–į¬Ľ –Ī—č–Ľ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ –ł –ĺ–Ņ–ł—Ā–į–Ĺ –≥–ĺ–Ľ–Ľ–į–Ĺ–ī—Ā–ļ–ł–ľ–ł —Ö–ł–ľ–ł–ļ–į–ľ–ł –Į.–†. –Ē–Ķ–Ļ–ľ–į–Ĺ–ĺ–ľ[–į–Ĺ–≥–Ľ.], –ü–ĺ—ā—Ā-–≤–į–Ĺ-–Ę—Ä–ĺ–ĺ—Ā—ā–≤–ł–ļ–ĺ–ľ, –Ď–ĺ–Ĺ–ī–ĺ–ľ –ł –õ–į—É–≤–Ķ—Ä–Ķ–Ĺ–Ī—É—Ä–≥–ĺ–ľ –≤ 1795 –≥–ĺ–ī—É. –ě–Ĺ–ł –Ĺ–į–∑–≤–į–Ľ–ł –Ķ–≥–ĺ ¬ę–ľ–į—Ā–Ľ–ĺ—Ä–ĺ–ī–Ĺ—č–ľ –≥–į–∑–ĺ–ľ¬Ľ —ā–į–ļ –ļ–į–ļ –Ņ—Ä–ł –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł–ł —Ā —Ö–Ľ–ĺ—Ä–ĺ–ľ, –ĺ–Ĺ –ĺ–Ī—Ä–į–∑–ĺ–≤—č–≤–į–Ľ –ľ–į—Ā–Ľ—Ź–Ĺ–ł—Ā—ā—É—é –∂–ł–ī–ļ–ĺ—Ā—ā—Ć ‚ÄĒ –ī–ł—Ö–Ľ–ĺ—Ä—ć—ā–į–Ĺ (–ĺ–Ī —ć—ā–ĺ–ľ —Ā—ā–į–Ľ–ĺ –ł–∑–≤–Ķ—Ā—ā–Ĺ–ĺ –Ņ–ĺ–∑–ī–Ĺ–Ķ–Ķ). –ü–ĺ-—Ą—Ä–į–Ĺ—Ü—É–∑—Ā–ļ–ł ¬ę–ľ–į—Ā–Ľ–ĺ—Ä–ĺ–ī–Ĺ—č–Ļ¬Ľ ‚ÄĒ ol√©fiant. –§—Ä–į–Ĺ—Ü—É–∑—Ā–ļ–ł–Ļ —Ö–ł–ľ–ł–ļ –ź–Ĺ—ā—É–į–Ĺ –§—É—Ä–ļ—Ä—É–į –≤–≤—Ď–Ľ —ć—ā–ĺ—ā —ā–Ķ—Ä–ľ–ł–Ĺ –≤ –Ņ—Ä–į–ļ—ā–ł–ļ—É, –į –ļ–ĺ–≥–ī–į –Ī—č–Ľ–ł –ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ—č –ī—Ä—É–≥–ł–Ķ —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī—č —ā–į–ļ–ĺ–≥–ĺ –∂–Ķ —ā–ł–Ņ–į, —ć—ā–ĺ –Ĺ–į–∑–≤–į–Ĺ–ł–Ķ —Ā—ā–į–Ľ–ĺ –ĺ–Ī—Č–ł–ľ –ī–Ľ—Ź –≤—Ā–Ķ–≥–ĺ –ļ–Ľ–į—Ā—Ā–į –ĺ–Ľ–Ķ—Ą–ł–Ĺ–ĺ–≤ (–ł–Ľ–ł, –Ņ–ĺ —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ĺ–ĺ–ľ–Ķ–Ĺ–ļ–Ľ–į—ā—É—Ä–Ķ, –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤)[5].
–í –Ĺ–į—á–į–Ľ–Ķ XIX –≤–Ķ–ļ–į —Ą—Ä–į–Ĺ—Ü—É–∑—Ā–ļ–ł–Ļ —Ö–ł–ľ–ł–ļ –Ė. –ď–Ķ–Ļ-–õ—é—Ā—Ā–į–ļ –ĺ–Ī–Ĺ–į—Ä—É–∂–ł–Ľ, —á—ā–ĺ —ć—ā–į–Ĺ–ĺ–Ľ —Ā–ĺ—Ā—ā–ĺ–ł—ā –ł–∑ ¬ę–ľ–į—Ā–Ľ–ĺ—Ä–ĺ–ī–Ĺ–ĺ–≥–嬼 –≥–į–∑–į –ł –≤–ĺ–ī—č. –≠—ā–ĺ—ā –∂–Ķ –≥–į–∑ –ĺ–Ĺ –ĺ–Ī–Ĺ–į—Ä—É–∂–ł–Ľ –ł –≤ —Ö–Ľ–ĺ—Ä–ł—Ā—ā–ĺ–ľ —ć—ā–ł–Ľ–Ķ[6]. –í 1828 –≥–ĺ–ī—É –Ė. –Ē—é–ľ–į –ł –ü. –Ď—É–Ľ–Ľ–Ķ–Ļ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–ł–Ľ–ł, —á—ā–ĺ —ć—ā–ł–Ľ–Ķ–Ĺ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź–Ķ—ā —Ā–ĺ–Ī–ĺ–Ļ –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–Ķ, —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ–Ķ –ī–į–≤–į—ā—Ć —Ā–ĺ–Ľ–ł –Ņ–ĺ–ī–ĺ–Ī–Ĺ–ĺ –į–ľ–ľ–ł–į–ļ—É. –Į–ļ–ĺ–Ī –Ď–Ķ—Ä—Ü–Ķ–Ľ–ł—É—Ā –Ņ—Ä–ł–Ĺ—Ź–Ľ —ć—ā—É –ł–ī–Ķ—é, –Ĺ–į–∑–≤–į–≤ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ ¬ę—ć—ā–Ķ—Ä–ł–Ĺ–ĺ–ľ¬Ľ –ł –ĺ–Ī–ĺ–∑–Ĺ–į—á–ł–≤ –Ī—É–ļ–≤–ĺ–Ļ E[7].
–ě–Ņ—Ä–Ķ–ī–Ķ–Ľ–ł–≤, —á—ā–ĺ —ć—ā–ł–Ľ–Ķ–Ĺ —Ā–ĺ—Ā—ā–ĺ–ł—ā –ł–∑ –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į –ł —É–≥–Ľ–Ķ—Ä–ĺ–ī–į, –ī–ĺ–Ľ–≥–ĺ–Ķ –≤—Ä–Ķ–ľ—Ź —Ö–ł–ľ–ł–ļ–ł –Ĺ–Ķ –ľ–ĺ–≥–Ľ–ł –≤—č–Ņ–ł—Ā–į—ā—Ć –Ķ–≥–ĺ –Ĺ–į—Ā—ā–ĺ—Ź—Č—É—é —Ą–ĺ—Ä–ľ—É–Ľ—É. –í 1848 –≥–ĺ–ī—É –ö–ĺ–Ľ—Ć–Ī–Ķ –Ņ–ł—Ā–į–Ľ —Ą–ĺ—Ä–ľ—É–Ľ—É —ć—ā–ł–Ľ–Ķ–Ĺ–į –ļ–į–ļ –°4–Ě4, —ć—ā–ĺ–≥–ĺ –∂–Ķ –ľ–Ĺ–Ķ–Ĺ–ł—Ź –Ņ—Ä–ł–ī–Ķ—Ä–∂–ł–≤–į–Ľ—Ā—Ź –ł –õ–ł–Ī–ł—Ö. –Ė. –Ē—é–ľ–į –Ņ—Ä–į–≤–ł–Ľ—Ć–Ĺ–ĺ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–ł–Ľ —Ā–ĺ—Ā—ā–į–≤ –≤–Ķ—Č–Ķ—Ā—ā–≤–į, –Ĺ–ĺ –Ķ–≥–ĺ —Ā—ā—Ä—É–ļ—ā—É—Ä–į –Ņ–ĺ-–Ņ—Ä–Ķ–∂–Ĺ–Ķ–ľ—É –Ī—č–Ľ–į –ĺ–Ņ–ł—Ā–į–Ĺ–į –Ĺ–Ķ–≤–Ķ—Ä–Ĺ–ĺ: –°2–Ě–Ě3[6].
–í 1862 –≥–ĺ–ī—É –Ĺ–Ķ–ľ–Ķ—Ü–ļ–ł–Ļ —Ö–ł–ľ–ł–ļ-–ĺ—Ä–≥–į–Ĺ–ł–ļ –≠. –≠—Ä–Ľ–Ķ–Ĺ–ľ–Ķ–Ļ–Ķ—Ä –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–ł–Ľ –Ĺ–į–Ľ–ł—á–ł–Ķ –≤ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–Ķ —ć—ā–ł–Ľ–Ķ–Ĺ–į –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł, –į –≤ 1870 –≥–ĺ–ī—É –ł–∑–≤–Ķ—Ā—ā–Ĺ—č–Ļ —Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ł–Ļ —É—á—Ď–Ĺ—č–Ļ –ź. –ú. –Ď—É—ā–Ľ–Ķ—Ä–ĺ–≤ –Ņ—Ä–ł–∑–Ĺ–į–Ľ —ć—ā—É —ā–ĺ—á–ļ—É –∑—Ä–Ķ–Ĺ–ł—Ź –Ņ—Ä–į–≤–ł–Ľ—Ć–Ĺ–ĺ–Ļ, –Ņ–ĺ–ī—ā–≤–Ķ—Ä–ī–ł–≤ –Ķ—Ď –Ņ—Ä–ł—Ä–ĺ–ī—É —ć–ļ—Ā–Ņ–Ķ—Ä–ł–ľ–Ķ–Ĺ—ā–į–Ľ—Ć–Ĺ–ĺ[8].
–Ě–į—Ö–ĺ–∂–ī–Ķ–Ĺ–ł–Ķ –≤ –Ņ—Ä–ł—Ä–ĺ–ī–Ķ –ł —Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–į—Ź —Ä–ĺ–Ľ—Ć –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–í –Ņ—Ä–ł—Ä–ĺ–ī–Ķ –į—Ü–ł–ļ–Ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ—č –Ņ—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł –Ĺ–Ķ –≤—Ā—ā—Ä–Ķ—á–į—é—ā—Ā—Ź[9]. –ü—Ä–ĺ—Ā—ā–Ķ–Ļ—ą–ł–Ļ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–ł—ā–Ķ–Ľ—Ć —ć—ā–ĺ–≥–ĺ –ļ–Ľ–į—Ā—Ā–į –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ ‚ÄĒ —ć—ā–ł–Ľ–Ķ–Ĺ (C2H4) ‚ÄĒ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –≥–ĺ—Ä–ľ–ĺ–Ĺ–ĺ–ľ –ī–Ľ—Ź —Ä–į—Ā—ā–Ķ–Ĺ–ł–Ļ –ł –≤ –Ĺ–Ķ–∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–ľ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–Ķ –≤ –Ĺ–ł—Ö —Ā–ł–Ĺ—ā–Ķ–∑–ł—Ä—É–Ķ—ā—Ā—Ź.
–ě–ī–ł–Ĺ –ł–∑ –Ĺ–Ķ–ľ–Ĺ–ĺ–≥–ł—Ö –Ņ—Ä–ł—Ä–ĺ–ī–Ĺ—č—Ö –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ ‚ÄĒ –ľ—É—Ā–ļ–į–Ľ—É—Ä (—Ü–ł—Ā- —ā—Ä–ł–ļ–ĺ–∑–Ķ–Ĺ-9) —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ–ĺ–Ľ–ĺ–≤—č–ľ –į—ā—ā—Ä–į–ļ—ā–į–Ĺ—ā–ĺ–ľ —Ā–į–ľ–ļ–ł –ī–ĺ–ľ–į—ą–Ĺ–Ķ–Ļ –ľ—É—Ö–ł (Musca domestica).

–Ě–ł–∑—ą–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ—č –≤ –≤—č—Ā–ĺ–ļ–ł—Ö –ļ–ĺ–Ĺ—Ü–Ķ–Ĺ—ā—Ä–į—Ü–ł—Ź—Ö –ĺ–Ī–Ľ–į–ī–į—é—ā –Ĺ–į—Ä–ļ–ĺ—ā–ł—á–Ķ—Ā–ļ–ł–ľ —ć—Ą—Ą–Ķ–ļ—ā–ĺ–ľ. –í—č—Ā—ą–ł–Ķ —á–Ľ–Ķ–Ĺ—č —Ä—Ź–ī–į —ā–į–ļ–∂–Ķ –≤—č–∑—č–≤–į—é—ā —Ā—É–ī–ĺ—Ä–ĺ–≥–ł –ł —Ä–į–∑–ī—Ä–į–∂–Ķ–Ĺ–ł–Ķ —Ā–Ľ–ł–∑–ł—Ā—ā—č—Ö –ĺ–Ī–ĺ–Ľ–ĺ—á–Ķ–ļ –ī—č—Ö–į—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –Ņ—É—ā–Ķ–Ļ[10].
–ě—ā–ī–Ķ–Ľ—Ć–Ĺ—č–Ķ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–ł—ā–Ķ–Ľ–ł –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ :
–§–ł–∑–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į
- –Ę–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä—č –Ņ–Ľ–į–≤–Ľ–Ķ–Ĺ–ł—Ź –ł –ļ–ł–Ņ–Ķ–Ĺ–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ (—É–Ņ—Ä–ĺ—Č—Ď–Ĺ–Ĺ–ĺ) —É–≤–Ķ–Ľ–ł—á–ł–≤–į—é—ā—Ā—Ź —Ā –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–Ļ –ľ–į—Ā—Ā–ĺ–Ļ –ł –ī–Ľ–ł–Ĺ–ĺ–Ļ –≥–Ľ–į–≤–Ĺ–ĺ–Ļ —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–Ļ —Ü–Ķ–Ņ–ł.
- –ü—Ä–ł –Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ—č—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –į–Ľ–ļ–Ķ–Ĺ—č —Ā C2H4 –ī–ĺ C4H8 ‚ÄĒ –≥–į–∑—č; —Ā –Ņ–Ķ–Ĺ—ā–Ķ–Ĺ–į C5H10 –ī–ĺ –≥–Ķ–Ņ—ā–į–ī–Ķ—Ü–Ķ–Ĺ–į C17H34 –≤–ļ–Ľ—é—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ ‚ÄĒ –∂–ł–ī–ļ–ĺ—Ā—ā–ł, –į –Ĺ–į—á–ł–Ĺ–į—Ź —Ā –ĺ–ļ—ā–į–ī–Ķ—Ü–Ķ–Ĺ–į C18H36 ‚ÄĒ —ā–≤—Ď—Ä–ī—č–Ķ –≤–Ķ—Č–Ķ—Ā—ā–≤–į. –ź–Ľ–ļ–Ķ–Ĺ—č –Ĺ–Ķ —Ä–į—Ā—ā–≤–ĺ—Ä—Ź—é—ā—Ā—Ź –≤ –≤–ĺ–ī–Ķ, –Ĺ–ĺ —Ö–ĺ—Ä–ĺ—ą–ĺ —Ä–į—Ā—ā–≤–ĺ—Ä—Ź—é—ā—Ā—Ź –≤ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ä–į—Ā—ā–≤–ĺ—Ä–ł—ā–Ķ–Ľ—Ź—Ö.
| –§–ł–∑–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤.[11]
|
| ‚ĄĖ |
–Ě–į–∑–≤–į–Ĺ–ł–Ķ |
–§–ĺ—Ä–ľ—É–Ľ–į |
–Ę –Ņ–Ľ–į–≤–Ľ–Ķ–Ĺ–ł—Ź, ¬į C |
–Ę –ļ–ł–Ņ–Ķ–Ĺ–ł—Ź, ¬į C |
–ü–Ľ–ĺ—ā–Ĺ–ĺ—Ā—ā—Ć, d20
4
|
| 1 |
–≠—ā–ł–Ľ–Ķ–Ĺ |
–°2H4 |
‚ąí169,1 |
‚ąí103,7 |
0,5700*
|
| 2 |
–ü—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ |
C3H6 |
‚ąí187,6 |
‚ąí47,7 |
0,5193*
|
| 3 |
–Ď—É—ā–Ķ–Ĺ-1 |
C4H8 |
‚ąí185,3 |
‚ąí6,3 |
0,5951*
|
| 4 |
—Ü–ł—Ā-–Ď—É—ā–Ķ–Ĺ-2 |
CH3-CH=CH-CH3 |
‚ąí138,9 |
3,7 |
0,6213
|
| 5 |
—ā—Ä–į–Ĺ—Ā-–Ď—É—ā–Ķ–Ĺ-2 |
CH3-CH=CH-CH3 |
‚ąí105,5 |
0,9 |
0,6042
|
| 6 |
2-–ú–Ķ—ā–ł–Ľ–Ņ—Ä–ĺ–Ņ–Ķ–Ĺ-1 |
CH3-C(CH3)=CH2 |
‚ąí140,4 |
‚ąí7,0 |
0,5942*
|
| 7 |
–ü–Ķ–Ĺ—ā–Ķ–Ĺ-1 |
CH2=CH-CH2-CH2-CH3 |
‚ąí165,2 |
30,1 |
0,6405
|
| 8 |
–ď–Ķ–ļ—Ā–Ķ–Ĺ-1 |
CH2=CH-CH2-CH2-CH2-CH3 |
‚ąí139,8 |
63,5 |
0,6730
|
| 9 |
–ď–Ķ–Ņ—ā–Ķ–Ĺ-1[–į–Ĺ–≥–Ľ.] |
–°7H14 |
‚ąí119,0 |
93,6 |
0,6970
|
| 10 |
–ě–ļ—ā–Ķ–Ĺ-1 |
–°8H16 |
‚ąí101,7 |
121,3 |
0,7140
|
| … |
–ď–Ķ–Ņ—ā–į–ī–Ķ—Ü–Ķ–Ĺ[12] |
–°17H34 |
4,1 |
284,4 |
0,7811
|
* –ó–Ĺ–į—á–Ķ–Ĺ–ł—Ź –ł–∑–ľ–Ķ—Ä–Ķ–Ĺ—č –Ņ—Ä–ł —ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–Ķ –ļ–ł–Ņ–Ķ–Ĺ–ł—Ź.
–•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į
–ź–Ľ–ļ–Ķ–Ĺ—č —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł –į–ļ—ā–ł–≤–Ĺ—č. –ė—Ö —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į –≤–ĺ –ľ–Ĺ–ĺ–≥–ĺ–ľ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź—é—ā—Ā—Ź –Ĺ–į–Ľ–ł—á–ł–Ķ–ľ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł.
–Ē–Ľ—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ—č —Ä–Ķ–į–ļ—Ü–ł–ł —ć–Ľ–Ķ–ļ—ā—Ä–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ł —Ä–Ķ–į–ļ—Ü–ł–ł —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź. –†–Ķ–į–ļ—Ü–ł–ł –Ĺ—É–ļ–Ľ–Ķ–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ĺ–Ī—č—á–Ĺ–ĺ —ā—Ä–Ķ–Ī—É—é—ā –Ĺ–į–Ľ–ł—á–ł–Ķ —Ā–ł–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ĺ—É–ļ–Ľ–Ķ–ĺ—Ą–ł–Ľ–į –ł –ī–Ľ—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –Ĺ–Ķ —ā–ł–Ņ–ł—á–Ĺ—č.
–ě—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ć—é –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ —Ź–≤–Ľ—Ź—é—ā—Ā—Ź —ā–į–ļ–∂–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł —Ü–ł–ļ–Ľ–ĺ–Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ł –ľ–Ķ—ā–į—ā–Ķ–∑–ł—Ā–į.
–ź–Ľ–ļ–Ķ–Ĺ—č –Ľ–Ķ–≥–ļ–ĺ –≤—Ā—ā—É–Ņ–į—é—ā –≤ —Ä–Ķ–į–ļ—Ü–ł–ł –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł—Ź, –≥–ł–ī—Ä–ł—Ä—É—é—ā—Ā—Ź —Ā —Ā–ł–Ľ—Ć–Ĺ—č–ľ–ł –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–ł—ā–Ķ–Ľ—Ź–ľ–ł –ł–Ľ–ł –≤–ĺ–ī–ĺ—Ä–ĺ–ī–ĺ–ľ –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–ĺ–≤, –į —ā–į–ļ–∂–Ķ —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ—č –ļ —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–ľ—É –∑–į–ľ–Ķ—Č–Ķ–Ĺ–ł—é.
–†–Ķ–į–ļ—Ü–ł–ł —ć–Ľ–Ķ–ļ—ā—Ä–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź
–í –ī–į–Ĺ–Ĺ—č—Ö —Ä–Ķ–į–ļ—Ü–ł—Ź—Ö –į—ā–į–ļ—É—é—Č–Ķ–Ļ —á–į—Ā—ā–ł—Ü–Ķ–Ļ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —ć–Ľ–Ķ–ļ—ā—Ä–ĺ—Ą–ł–Ľ.
–ď–į–Ľ–ĺ–≥–Ķ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤, –Ņ—Ä–ĺ—Ö–ĺ–ī—Ź—Č–Ķ–Ķ –≤ –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ –ł–Ĺ–ł—Ü–ł–į—ā–ĺ—Ä–ĺ–≤ —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ—č—Ö —Ä–Ķ–į–ļ—Ü–ł–Ļ ‚ÄĒ —ā–ł–Ņ–ł—á–Ĺ–į—Ź —Ä–Ķ–į–ļ—Ü–ł—Ź —ć–Ľ–Ķ–ļ—ā—Ä–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź. –ě–Ĺ–į –Ņ—Ä–ĺ–≤–ĺ–ī–ł—ā—Ā—Ź –≤ —Ā—Ä–Ķ–ī–Ķ –Ĺ–Ķ–Ņ–ĺ–Ľ—Ź—Ä–Ĺ—č—Ö –ł–Ĺ–Ķ—Ä—ā–Ĺ—č—Ö —Ä–į—Ā—ā–≤–ĺ—Ä–ł—ā–Ķ–Ľ–Ķ–Ļ (–Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä: CCl4):

–†–Ķ–į–ļ—Ü–ł—Ź –≥–į–Ľ–ĺ–≥–Ķ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ā—ā–Ķ—Ä–Ķ–ĺ—Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ĺ–į ‚ÄĒ- –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā —Ā –Ņ—Ä–ĺ—ā–ł–≤–ĺ–Ņ–ĺ–Ľ–ĺ–∂–Ĺ—č—Ö —Ā—ā–ĺ—Ä–ĺ–Ĺ –ĺ—ā–Ĺ–ĺ—Ā–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –Ņ–Ľ–ĺ—Ā–ļ–ĺ—Ā—ā–ł –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—č –į–Ľ–ļ–Ķ–Ĺ–į[2]
–ú–Ķ—Ö–į–Ĺ–ł–∑–ľ —Ä–Ķ–į–ļ—Ü–ł–Ļ –Ņ–ĺ–ī–ĺ–Ī–Ĺ–ĺ–≥–ĺ —ā–ł–Ņ–į –≤ –ĺ–Ī—Č–Ķ–ľ –≤–ł–ī–Ķ:

–≠–Ľ–Ķ–ļ—ā—Ä–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–Ķ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –≥–į–Ľ–ĺ–≥–Ķ–Ĺ–≤–ĺ–ī–ĺ—Ä–ĺ–ī–ĺ–≤ –ļ –į–Ľ–ļ–Ķ–Ĺ–į–ľ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ–ĺ –Ņ—Ä–į–≤–ł–Ľ—É –ú–į—Ä–ļ–ĺ–≤–Ĺ–ł–ļ–ĺ–≤–į:

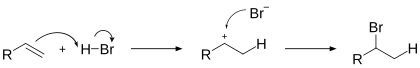
–ě–ī–Ĺ–į–ļ–ĺ –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł –Ņ–Ķ—Ä–Ķ–ļ–ł—Ā–Ķ–Ļ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –Ņ—Ä–ĺ—ā–ł–≤ —ć—ā–ĺ–≥–ĺ –Ņ—Ä–į–≤–ł–Ľ–į (—ć—Ą—Ą–Ķ–ļ—ā –•–į—Ä–į—ą–į)[2]:

–≠—ā–ĺ –ĺ–Ī—ä—Ź—Ā–Ĺ—Ź–Ķ—ā—Ā—Ź —ā–Ķ–ľ, —á—ā–ĺ —Ä–Ķ–į–ļ—Ü–ł—Ź –≤ –ī–į–Ĺ–Ĺ–ĺ–ľ —Ā–Ľ—É—á–į–Ķ –Ī—É–ī–Ķ—ā –Ņ—Ä–ĺ—ā–Ķ–ļ–į—ā—Ć –Ņ–ĺ —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–ľ—É –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—É –ł –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ —Ä–į–ī–ł–ļ–į–Ľ–į Br. –ł–ī—Ď—ā –Ņ–ĺ —Ā—ā–Ķ—Ä–ł—á–Ķ—Ā–ļ–ł –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –ī–ĺ—Ā—ā—É–Ņ–Ĺ–ĺ–ľ—É –ļ–ĺ–Ĺ—Ü–Ķ–≤–ĺ–ľ—É –į—ā–ĺ–ľ—É —É–≥–Ľ–Ķ—Ä–ĺ–ī–į –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł:

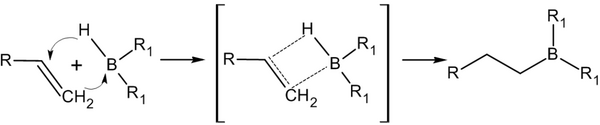
–ü—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –≥–ł–ī—Ä–ł–ī–ĺ–≤ –Ī–ĺ—Ä–į –ļ –į–Ľ–ļ–Ķ–Ĺ–į–ľ –ł –Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–Ķ–Ķ –ł—Ö —Ä–į—Ā—Č–Ķ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ –≤ —Č–Ķ–Ľ–ĺ—á–Ĺ–ĺ–Ļ —Ā—Ä–Ķ–ī–Ķ, –ĺ—ā–ļ—Ä—č—ā–ĺ–Ķ –ď. –Ď—Ä–į—É–Ĺ–ĺ–ľ –≤ 1958 –≥–ĺ–ī—É, —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā—ā–ĺ–Ľ—Ć –≤–į–∂–Ĺ–ĺ–Ļ —Ä–Ķ–į–ļ—Ü–ł–Ķ–Ļ, —á—ā–ĺ –∑–į –Ķ—Ď –ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ–ł–Ķ –ł –ł–∑—É—á–Ķ–Ĺ–ł–Ķ –≤ 1979 –≥–ĺ–ī—É —É—á—Ď–Ĺ—č–Ļ –Ī—č–Ľ —É–ī–ĺ—Ā—ā–ĺ–Ķ–Ĺ –Ě–ĺ–Ī–Ķ–Ľ–Ķ–≤—Ā–ļ–ĺ–Ļ –Ņ—Ä–Ķ–ľ–ł–ł –Ņ–ĺ —Ö–ł–ľ–ł–ł[13].
–ü—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –ľ–Ĺ–ĺ–≥–ĺ—Ā—ā—É–Ņ–Ķ–Ĺ—á–į—ā–ĺ —Ā –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –Ņ—Ä–ĺ–ľ–Ķ–∂—É—ā–ĺ—á–Ĺ–ĺ–≥–ĺ —Ü–ł–ļ–Ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –į–ļ—ā–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā–į, –Ņ—Ä–ł—á—Ď–ľ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –Ī–ĺ—Ä–į –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ—Ä–ĺ—ā–ł–≤ –Ņ—Ä–į–≤–ł–Ľ–į –ú–į—Ä–ļ–ĺ–≤–Ĺ–ł–ļ–ĺ–≤–į ‚ÄĒ –ļ –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –≥–ł–ī—Ä–ĺ–≥–Ķ–Ĺ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–ľ—É –į—ā–ĺ–ľ—É —É–≥–Ľ–Ķ—Ä–ĺ–ī–į:
–í —Ā–ł–Ĺ—ā–Ķ–∑–Ķ –ĺ–Ī—č—á–Ĺ–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ—ā—Ā—Ź –Ĺ–Ķ —Ā–į–ľ –ī–ł–Ī–ĺ—Ä–į–Ĺ, –į –Ķ–≥–ĺ –ī–ĺ–Ĺ–ĺ—Ä–Ĺ–ĺ-–į–ļ—Ü–Ķ–Ņ—ā–ĺ—Ä–Ĺ—č–Ļ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā —Ā –Ņ—Ä–ĺ—Ā—ā—č–ľ —ć—Ą–ł—Ä–ĺ–ľ:

–ź–Ľ–ļ–ł–Ľ–Ī–ĺ—Ä–į–Ĺ—č –Ľ–Ķ–≥–ļ–ĺ —Ä–į—Ā—Č–Ķ–Ņ–Ľ—Ź—é—ā—Ā—Ź. –Ę–į–ļ –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī–į –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į –≤ —Č–Ķ–Ľ–ĺ—á–Ĺ–ĺ–Ļ —Ā—Ä–Ķ–ī–Ķ –ĺ–Ī—Ä–į–∑—É—é—ā—Ā—Ź —Ā–Ņ–ł—Ä—ā—č:

–†–Ķ–į–ļ—Ü–ł—Ź –≥–ł–ī—Ä–ĺ–Ī–ĺ—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ä–Ķ–į–ļ—Ü–ł–Ķ–Ļ —Ā–ł–Ĺ-–Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź ‚ÄĒ –Ķ—Ď —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–ľ —Ā—ā–į–Ĺ–ĺ–≤—Ź—ā—Ā—Ź —Ü–ł—Ā-–į–ī–ī—É–ļ—ā—č.
–ď–ł–ī—Ä–į—ā–į—Ü–ł—Ź
–†–Ķ–į–ļ—Ü–ł—Ź –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –≤–ĺ–ī—č –ļ –į–Ľ–ļ–Ķ–Ĺ–į–ľ –Ņ—Ä–ĺ—ā–Ķ–ļ–į–Ķ—ā –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł —Ā–Ķ—Ä–Ĺ–ĺ–Ļ –ļ–ł—Ā–Ľ–ĺ—ā—č[14]:

–†–Ķ–į–ļ—Ü–ł—Ź –Ņ—Ä–ĺ—ā–Ķ–ļ–į–Ķ—ā –Ņ–ĺ –Ņ—Ä–į–≤–ł–Ľ—É –ú–į—Ä–ļ–ĺ–≤–Ĺ–ł–ļ–ĺ–≤–į.
–ü—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –į–Ľ–ļ–į–Ĺ–ĺ–≤ –ļ –į–Ľ–ļ–Ķ–Ĺ–į–ľ –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł –ļ–ł—Ā–Ľ–ĺ—ā–Ĺ–ĺ–≥–ĺ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į (HF –ł–Ľ–ł H2SO4) –Ņ—Ä–ł –Ĺ–ł–∑–ļ–ł—Ö —ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–į—Ö –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—é —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī–į —Ā –Ī–ĺ–Ľ—Ć—ą–Ķ–Ļ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–Ļ –ľ–į—Ā—Ā–ĺ–Ļ –ł —á–į—Ā—ā–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ—ā—Ā—Ź –≤ –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł[15]:

–Ē–į–Ĺ–Ĺ–į—Ź —Ä–Ķ–į–ļ—Ü–ł—Ź —ā–į–ļ–∂–Ķ –ľ–ĺ–∂–Ķ—ā –Ņ—Ä–ĺ—ā–Ķ–ļ–į—ā—Ć –Ņ–ĺ —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ–ĺ—Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–ľ—É –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—É –≤ –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į –Ņ—Ä–ł –≤—č—Ā–ĺ–ļ–ĺ–Ļ —ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–Ķ (500 ¬įC) –ł –ī–į–≤–Ľ–Ķ–Ĺ–ł–ł (15-30 –ú–ü–į)[14].
–ü—Ä–ĺ—á–ł–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł —ć–Ľ–Ķ–ļ—ā—Ä–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź
–Ē–Ľ—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ —ā–į–ļ–∂–Ķ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ—č —Ā–Ľ–Ķ–ī—É—é—Č–ł–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł —ć–Ľ–Ķ–ļ—ā—Ä–ĺ—Ą–ł–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź[14]:

- –ü–ĺ–Ľ—É—á–Ķ–Ĺ–ł–Ķ —Ā–Ņ–ł—Ä—ā–ĺ–≤ –Ņ–ĺ —Ä–Ķ–į–ļ—Ü–ł–ł –ĺ–ļ—Ā–ł–ľ–Ķ—Ä–ļ—É—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź-–ī–Ķ–ľ–Ķ—Ä–ļ—É—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź:




–†–Ķ–į–ļ—Ü–ł–ł —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź
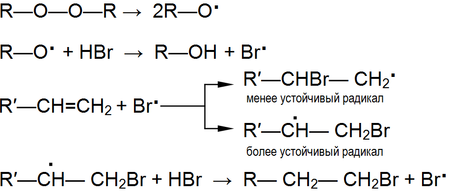
–í —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö, —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É—é—Č–ł—Ö –≥–ĺ–ľ–ĺ–Ľ–ł—ā–ł—á–Ķ—Ā–ļ–ĺ–ľ—É —Ä–į–∑—Ä—č–≤—É —Ā–≤—Ź–∑–ł, (–≤—č—Ā–ĺ–ļ–į—Ź —ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–į, –ĺ–Ī–Ľ—É—á–Ķ–Ĺ–ł–Ķ, –Ĺ–į–Ľ–ł—á–ł–Ķ —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ—č—Ö —Ä–į–ī–ł–ļ–į–Ľ–ĺ–≤ –ł –Ņ—Ä.) –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –ļ –į–Ľ–ļ–Ķ–Ĺ–į–ľ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ–ĺ —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–ľ—É –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—É[17].
 –Ņ–ĺ –Ņ—Ä–į–≤–ł–Ľ—É –ú–į—Ä–ļ–ĺ–≤–Ĺ–ł–ļ–ĺ–≤–į.
–Ņ–ĺ –Ņ—Ä–į–≤–ł–Ľ—É –ú–į—Ä–ļ–ĺ–≤–Ĺ–ł–ļ–ĺ–≤–į.


–ł —ā. –Ņ.
–ú–Ķ—Ö–į–Ĺ–ł–∑–ľ —Ä–Ķ–į–ļ—Ü–ł–ł:
–†–Ķ–į–ļ—Ü–ł–ł –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ļ–į—Ä–Ī–Ķ–Ĺ–ĺ–≤
–ö–į—Ä–Ī–Ķ–Ĺ—č CR2: ‚ÄĒ –≤—č—Ā–ĺ–ļ–ĺ—Ä–Ķ–į–ļ—Ü–ł–ĺ–Ĺ–Ĺ—č–Ķ –ļ–ĺ—Ä–ĺ—ā–ļ–ĺ–∂–ł–≤—É—Č–ł–Ķ —á–į—Ā—ā–ł—Ü—č, –ļ–ĺ—ā–ĺ—Ä—č–Ķ —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ—č –Ľ–Ķ–≥–ļ–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ—Ź—ā—Ć—Ā—Ź –ļ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤[18]. –í —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ļ–į—Ä–Ī–Ķ–Ĺ–į –ĺ–Ī—Ä–į–∑—É—é—ā—Ā—Ź –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č–Ķ —Ü–ł–ļ–Ľ–ĺ–Ņ—Ä–ĺ–Ņ–į–Ĺ–į:

–ö–į—Ä–Ī–Ķ–Ĺ—č –≤ –Ī–ĺ–Ľ–Ķ–Ķ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ–ĺ–ľ –ī–Ľ—Ź –Ĺ–ł—Ö —Ā–ł–Ĺ–≥–Ľ–Ķ—ā–Ĺ–ĺ–ľ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–ł, –≤—Ā—ā—É–Ņ–į—Ź –≤ —Ä–Ķ–į–ļ—Ü–ł—é, –ī–į—é—ā —Ā—ā–Ķ—Ä–Ķ–ĺ—Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ĺ—č–Ķ –Ņ—Ä–ĺ–ī—É–ļ—ā—č —Ā–ł–Ĺ-–Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź[14].
–ü–ĺ–ľ–ł–ľ–ĺ —Ā–ĺ–Ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –ļ–į—Ä–Ī–Ķ–Ĺ–į, –≤ –Ņ–ĺ–ī–ĺ–Ī–Ĺ—č–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł –ľ–ĺ–≥—É—ā –≤—Ā—ā—É–Ņ–į—ā—Ć –ł –Ķ–≥–ĺ –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č–Ķ[14]:
 –ł –Ņ—Ä.
–ł –Ņ—Ä.
–ß–į—Ā—ā–ĺ —Ä–Ķ–į–ļ—Ü–ł–ł –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ļ–į—Ä–Ī–Ķ–Ĺ–ĺ–≤ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī—Ź—ā –Ī–Ķ–∑ –Ņ—Ä—Ź–ľ—č—Ö –ī–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć—Ā—ā–≤ –ł—Ö —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł—Ź, —ā–ĺ –Ķ—Ā—ā—Ć –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā –ļ–į—Ä–Ī–Ķ–Ĺ–į. –Ē–Ľ—Ź —ć—ā–ĺ–≥–ĺ —Ā–Ľ—É—á–į—Ź, –į —ā–į–ļ–∂–Ķ –Ķ—Ā–Ľ–ł –≥–Ķ–Ĺ–Ķ—Ä–į—Ü–ł—Ź —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ–ĺ–≥–ĺ –ļ–į—Ä–Ī–Ķ–Ĺ–į —Ā—ā–į–≤–ł—ā—Ā—Ź –Ņ–ĺ–ī —Ā–ĺ–ľ–Ĺ–Ķ–Ĺ–ł–Ķ, –Ņ–ĺ–Ľ—Ć–∑—É—é—ā—Ā—Ź —ā–Ķ—Ä–ľ–ł–Ĺ–ĺ–ľ –ļ–į—Ä–Ī–Ķ–Ĺ–ĺ–ł–ī[19].
–í –Ľ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–Ĺ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ —á–į—Ā—ā–ĺ –Ņ–ĺ–Ľ—Ć–∑—É—é—ā—Ā—Ź —Ä–Ķ–į–ļ—Ü–ł–Ķ–Ļ –°–ł–ľ–ľ–ĺ–Ĺ—Ā–į ‚ÄĒ –°–ľ–ł—ā–į[–į–Ĺ–≥–Ľ.][20]:

–ü–ĺ–ī—Ä–ĺ–Ī–Ĺ–Ķ–Ķ –ĺ –ľ–Ķ—ā–ĺ–ī–į—Ö –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –ļ–į—Ä–Ī–Ķ–Ĺ–ĺ–≤ —Ā–ľ. —Ā—ā–į—ā—Ć—é –ö–į—Ä–Ī–Ķ–Ĺ—č.
–ď–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ (—Ä–Ķ–į–ļ—Ü–ł—Ź –°–į–Ī–į—ā—Ć–Ķ ‚ÄĒ –°–į–Ĺ–ī–Ķ—Ä–į–Ĺ)

–ď–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –Ĺ–Ķ–Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –≤–ĺ–ī–ĺ—Ä–ĺ–ī–ĺ–ľ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā —ā–ĺ–Ľ—Ć–ļ–ĺ –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į.
–ď–Ķ—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ĺ—č–ľ–ł –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į–ľ–ł –≥–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ā–Ľ—É–∂–į—ā –Ņ–Ľ–į—ā–ł–Ĺ–į, –Ņ–į–Ľ–Ľ–į–ī–ł–Ļ, –Ĺ–ł–ļ–Ķ–Ľ—Ć
[21].
–ď–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ľ–ĺ–∂–Ĺ–ĺ –Ņ—Ä–ĺ–≤–ĺ–ī–ł—ā—Ć –ł –≤ –∂–ł–ī–ļ–ĺ–Ļ —Ą–į–∑–Ķ —Ā –≥–ĺ–ľ–ĺ–≥–Ķ–Ĺ–Ĺ—č–ľ–ł –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į–ľ–ł (–Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä: –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä –£–ł–Ľ–ļ–ł–Ĺ—Ā–ĺ–Ĺ–į ((C6H5)3P)3Rh Cl)[21].
–í –ļ–į—á–Ķ—Ā—ā–≤–Ķ —Ä–Ķ–į–≥–Ķ–Ĺ—ā–ĺ–≤ –≥–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –ľ–ĺ–≥—É—ā –≤—č—Ā—ā—É–Ņ–į—ā—Ć –ī–ł–ł–ľ–ł–ī[–į–Ĺ–≥–Ľ.] (NH=NH), –ī–ł–Ī–ĺ—Ä–į–Ĺ (B2H6) –ł –ī—Ä[22].
–†–Ķ–į–ļ—Ü–ł–ł —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –∑–į–ľ–Ķ—Č–Ķ–Ĺ–ł—Ź
–ü—Ä–ł –≤—č—Ā–ĺ–ļ–ł—Ö —ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–į—Ö (–Ī–ĺ–Ľ–Ķ–Ķ 400 ¬įC) —Ä–Ķ–į–ļ—Ü–ł–ł —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź, –Ĺ–ĺ—Ā—Ź—Č–ł–Ķ –ĺ–Ī—Ä–į—ā–ł–ľ—č–Ļ —Ö–į—Ä–į–ļ—ā–Ķ—Ä, –Ņ–ĺ–ī–į–≤–Ľ—Ź—é—ā—Ā—Ź.
–í —ć—ā–ĺ–ľ —Ā–Ľ—É—á–į–Ķ —Ā—ā–į–Ĺ–ĺ–≤–ł—ā—Ā—Ź –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–ľ –Ņ—Ä–ĺ–≤–Ķ—Ā—ā–ł –∑–į–ľ–Ķ—Č–Ķ–Ĺ–ł–Ķ –į—ā–ĺ–ľ–į –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į, –Ĺ–į—Ö–ĺ–ī—Ź—Č–Ķ–≥–ĺ—Ā—Ź –≤ –į–Ľ–Ľ–ł–Ľ—Ć–Ĺ–ĺ–ľ –Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł–ł –Ņ—Ä–ł —Ā–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł–ł –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł:

–†–Ķ–į–ļ—Ü–ł—Ź –Ĺ–ĺ—Ā–ł—ā —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ—č–Ļ —Ö–į—Ä–į–ļ—ā–Ķ—Ä –ł –Ņ—Ä–ĺ—ā–Ķ–ļ–į–Ķ—ā –į–Ĺ–į–Ľ–ĺ–≥–ł—á–Ĺ–ĺ —Ö–Ľ–ĺ—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł—é –į–Ľ–ļ–į–Ĺ–ĺ–≤.
–ź–Ľ–Ľ–ł–Ľ—Ć–Ĺ–ĺ–Ķ –Ī—Ä–ĺ–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ĺ–Ī—č—á–Ĺ–ĺ –Ņ—Ä–ĺ–≤–ĺ–ī—Ź—ā N-–Ī—Ä–ĺ–ľ—Ā—É–ļ—Ü–ł–Ĺ–ł–ľ–ł–ī–ĺ–ľ (—Ä–Ķ–į–ļ—Ü–ł—Ź –í–ĺ–Ľ—Ź ‚ÄĒ –¶–ł–≥–Ľ–Ķ—Ä–į)[23] –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł –Ņ–Ķ—Ä–Ķ–ļ–ł—Ā–ł –Ī–Ķ–Ĺ–∑–ĺ–ł–Ľ–į –≤ —Ā—Ä–Ķ–ī–Ķ —ā–Ķ—ā—Ä–į—Ö–Ľ–ĺ—Ä–ľ–Ķ—ā–į–Ĺ–į –ł–Ľ–ł –≤ –Ī–ł–Ĺ–į—Ä–Ĺ–ĺ–Ļ —Ā–ľ–Ķ—Ā–ł –ī–ł–ľ–Ķ—ā–ł–Ľ—Ā—É–Ľ—Ć—Ą–ĺ–ļ—Ā–ł–ī–į –ł –≤–ĺ–ī—č[21]:

–ě–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł–Ķ
–ě–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ľ–ĺ–∂–Ķ—ā –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā—Ć –≤ –∑–į–≤–ł—Ā–ł–ľ–ĺ—Ā—ā–ł –ĺ—ā —É—Ā–Ľ–ĺ–≤–ł–Ļ –ł –≤–ł–ī–ĺ–≤ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ä–Ķ–į–≥–Ķ–Ĺ—ā–ĺ–≤ –ļ–į–ļ —Ā —Ä–į–∑—Ä—č–≤–ĺ–ľ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł, —ā–į–ļ –ł —Ā —Ā–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł–Ķ–ľ —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ–ĺ–≥–ĺ —Ā–ļ–Ķ–Ľ–Ķ—ā–į.
–ě–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł–Ķ –Ĺ–Ķ–ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł–ľ–ł –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ—Ź–ľ–ł
- –í –ľ—Ź–≥–ļ–ł—Ö —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł–Ķ –Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–ĺ–ľ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –Ņ–ĺ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –ī–≤—É—Ö –≥–ł–ī—Ä–ĺ–ļ—Ā–ł–Ľ—Ć–Ĺ—č—Ö –≥—Ä—É–Ņ–Ņ[24]:

–Ě–į –Ņ–Ķ—Ä–≤–ĺ–ľ —ć—ā–į–Ņ–Ķ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –ĺ–ļ—Ā–ł–ī–į –ĺ—Ā–ľ–ł—Ź –ļ –į–Ľ–ļ–Ķ–Ĺ—É, –∑–į—ā–Ķ–ľ –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–ł—ā–Ķ–Ľ—Ź (Zn –ł–Ľ–ł NaHSO3) –ĺ–Ī—Ä–į–∑–ĺ–≤–į–≤—ą–ł–Ļ—Ā—Ź –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā –Ņ–Ķ—Ä–Ķ—Ö–ĺ–ī–ł—ā –ļ –ī–ł–ĺ–Ľ—É (–†–Ķ–į–ļ—Ü–ł—Ź –ö—Ä–ł–≥–Ķ).
–ź–Ĺ–į–Ľ–ĺ–≥–ł—á–Ĺ–ĺ —Ä–Ķ–į–ļ—Ü–ł—Ź –ł–ī—Ď—ā –≤ –Ĺ–Ķ–Ļ—ā—Ä–į–Ľ—Ć–Ĺ–ĺ–Ļ –ł–Ľ–ł —Ā–Ľ–į–Ī–ĺ—Č–Ķ–Ľ–ĺ—á–Ĺ–ĺ–Ļ —Ā—Ä–Ķ–ī–Ķ –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ KMnO4 (–†–Ķ–į–ļ—Ü–ł—Ź –í–į–≥–Ĺ–Ķ—Ä–į)[24]:

- –ü—Ä–ł –ī–Ķ–Ļ—Ā—ā–≤–ł–ł –Ĺ–į –į–Ľ–ļ–Ķ–Ĺ—č —Ā–ł–Ľ—Ć–Ĺ—č—Ö –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ–Ķ–Ļ (KMnO4 –ł–Ľ–ł K2Cr2O7 –≤ —Ā—Ä–Ķ–ī–Ķ –Ě2SO4) –Ņ—Ä–ł –Ĺ–į–≥—Ä–Ķ–≤–į–Ĺ–ł–ł –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā —Ä–į–∑—Ä—č–≤ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł:
![{\displaystyle {\mathsf {R\!\!-\!\!CH\!\!=\!\!CH\!\!-\!\!R+[O]}}\rightarrow {\mathsf {2R\!\!-\!\!COOH}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/5848e6203a9341c8ca9a8d6a7251d29c949dfdf5)
![{\displaystyle {\mathsf {R\!\!-\!\!CH\!\!=\!\!C(R)(R')+[O]}}\rightarrow {\mathsf {R\!\!-\!\!COOH+R\!\!-\!\!C(O)\!\!-\!\!R'}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ea6c33e33f2be61289db42c78b1747f6409c7d50) (–ļ–Ķ—ā–ĺ–Ĺ)
(–ļ–Ķ—ā–ĺ–Ĺ)
- –Ě–Ķ–ļ–ĺ—ā–ĺ—Ä—č–Ķ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ–ł, –Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä –Ĺ–ł—ā—Ä–į—ā (III) —ā–į–Ľ–Ľ–ł—Ź, –ĺ–ļ–ł—Ā–Ľ—Ź—é—ā –į–Ľ–ļ–Ķ–Ĺ—č —Ā –Ņ–Ķ—Ä–Ķ–≥—Ä—É–Ņ–Ņ–ł—Ä–ĺ–≤–ļ–ĺ–Ļ –Ņ–ĺ —Ā–Ľ–Ķ–ī—É—é—Č–Ķ–Ļ —Ā—Ö–Ķ–ľ–Ķ[24]:
![{\displaystyle {\mathsf {R\!\!-\!\!C(R)\!\!=\!\!CH\!\!-\!\!R+[O]}}\rightarrow {\mathsf {R\!\!-\!\!C(O)\!\!-\!\!CH(R)R}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ea802883018bd47662e62ca08a22b0db107be036)
–ě–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł–Ķ –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł —Ā–ĺ–Ľ–Ķ–Ļ –Ņ–į–Ľ–Ľ–į–ī–ł—Ź
–í –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł —Ā–ĺ–Ľ–Ķ–Ļ –Ņ–į–Ľ–Ľ–į–ī–ł—Ź —ć—ā–ł–Ľ–Ķ–Ĺ –ĺ–ļ–ł—Ā–Ľ—Ź–Ķ—ā—Ā—Ź –ī–ĺ –į—Ü–Ķ—ā–į–Ľ—Ć–ī–Ķ–≥–ł–ī–į[2]:

–†–Ķ–į–ļ—Ü–ł—Ź –ł–ī—Ď—ā –≤ –ļ–ł—Ā–Ľ–ĺ–Ļ —Ā—Ä–Ķ–ī–Ķ –ł —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ—č–ľ —Ā–Ņ–ĺ—Ā–ĺ–Ī–ĺ–ľ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –į—Ü–Ķ—ā–į–Ľ—Ć–ī–Ķ–≥–ł–ī–į.
–ź–Ĺ–į–Ľ–ĺ–≥–ł—á–Ĺ–ĺ –ĺ–Ī—Ä–į–∑—É–Ķ—ā—Ā—Ź –į—Ü–Ķ—ā–ĺ–Ĺ –ł–∑ –Ņ—Ä–ĺ–Ņ–Ķ–Ĺ–į.
–≠–Ņ–ĺ–ļ—Ā–ł–ī–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ
–ü—Ä–ł –ī–Ķ–Ļ—Ā—ā–≤–ł–ł –Ĺ–į –į–Ľ–ļ–Ķ–Ĺ—č –Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ļ–į—Ä–Ī–ĺ–Ĺ–ĺ–≤—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā –ĺ–Ī—Ä–į–∑—É—é—ā—Ā—Ź —ć–Ņ–ĺ–ļ—Ā–ł–ī—č (—Ä–Ķ–į–ļ—Ü–ł—Ź –ü—Ä–ł–Ľ–Ķ–∂–į–Ķ–≤–į)[25]:

–†–Ķ–į–ļ—Ü–ł—Ź —ć–Ņ–ĺ–ļ—Ā–ł–ī–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ—ā—Ā—Ź –ī–Ľ—Ź –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź —ć—ā–ł–Ľ–Ķ–Ĺ–ĺ–ļ—Ā–ł–ī–į. –ě–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ–Ķ–ľ –≤—č—Ā—ā—É–Ņ–į–Ķ—ā –ļ–ł—Ā–Ľ–ĺ—Ä–ĺ–ī –≤–ĺ–∑–ī—É—Ö–į; –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā –ł–ī—Ď—ā –Ĺ–į —Ā–Ķ—Ä–Ķ–Ī—Ä—Ź–Ĺ–ĺ–ľ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–Ķ –Ņ—Ä–ł 200‚ÄĒ250 ¬įC –Ņ–ĺ–ī –ī–į–≤–Ľ–Ķ–Ĺ–ł–Ķ–ľ.
–ě–∑–ĺ–Ĺ–ĺ–Ľ–ł–∑
–ě–∑–ĺ–Ĺ–ĺ–Ľ–ł–∑ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ĺ–Ī—č—á–Ĺ–ĺ –Ņ—Ä–ĺ–≤–ĺ–ī—Ź—ā –Ņ—Ä–ł –Ĺ–ł–∑–ļ–ł—Ö —ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–į—Ö (–ĺ—ā ‚ąí80 –ī–ĺ ‚ąí30 ¬įC) –≤ –ł–Ĺ–Ķ—Ä—ā–Ĺ–ĺ–ľ —Ä–į—Ā—ā–≤–ĺ—Ä–ł—ā–Ķ–Ľ–Ķ (–≥–Ķ–ļ—Ā–į–Ĺ, —ā–Ķ—ā—Ä–į—Ö–Ľ–ĺ—Ä–ľ–Ķ—ā–į–Ĺ, —Ö–Ľ–ĺ—Ä–ĺ—Ą–ĺ—Ä–ľ, —ć—ā–ł–Ľ–į—Ü–Ķ—ā–į—ā –ł –Ņ—Ä.). –Ě–Ķ–Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ķ –Ņ—Ä–ĺ–ī—É–ļ—ā—č –ĺ–∑–ĺ–Ĺ–ĺ–Ľ–ł–∑–į –Ĺ–Ķ –≤—č–ī–Ķ–Ľ—Ź—é—ā, –į –Ņ–ĺ–ī–≤–Ķ—Ä–≥–į—é—ā –ī–į–Ľ—Ć–Ĺ–Ķ–Ļ—ą–Ķ–ľ—É –≥–ł–ī—Ä–ĺ–Ľ–ł–∑—É, –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł—é –ł–Ľ–ł –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—é[24].
–Ě–į –Ņ–Ķ—Ä–≤–ĺ–Ļ —Ā—ā–į–ī–ł–ł –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ķ –ĺ–∑–ĺ–Ĺ–į —Ā –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ĺ–∑–ĺ–Ĺ–ł–ī–į. –Ē–į–Ľ–Ķ–Ķ –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–ł—ā–Ķ–Ľ—Ź (–Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä: Zn + CH3COOH) –ĺ–∑–ĺ–Ĺ–ł–ī —Ä–į–∑–Ľ–į–≥–į–Ķ—ā—Ā—Ź:

–ē—Ā–Ľ–ł –≤–∑—Ź—ā—Ć –Ī–ĺ–Ľ–Ķ–Ķ —Ā–ł–Ľ—Ć–Ĺ—č–Ļ –≤–ĺ—Ā—Ā—ā–į–Ĺ–ĺ–≤–ł—ā–Ķ–Ľ—Ć, —Ā–ļ–į–∂–Ķ–ľ ‚ÄĒ –į–Ľ—é–ľ–ĺ–≥–ł–ī—Ä–ł–ī –Ľ–ł—ā–ł—Ź, –Ņ—Ä–ĺ–ī—É–ļ—ā–ĺ–ľ —Ä–Ķ–į–ļ—Ü–ł–ł –Ī—É–ī—É—ā —Ā–Ņ–ł—Ä—ā—č.

–í –ī–į–Ĺ–Ĺ–ĺ–ľ —Ā–Ľ—É—á–į–Ķ —Ä–į–∑–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ –ĺ–∑–ĺ–Ĺ–ł–ī–į –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –ĺ–ļ–ł—Ā–Ľ–ł—ā–Ķ–Ľ–Ķ–Ļ (–Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į, –ĺ–ļ—Ā–ł–ī —Ā–Ķ—Ä–Ķ–Ī—Ä–į, –Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ļ–ł—Ā–Ľ–ĺ—ā—č –ł –Ņ—Ä.[24]).
–ź–Ľ–ļ–Ķ–Ĺ—č –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į, –≤—č—Ā–ĺ–ļ–ĺ–Ļ —ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä—č –ł –ī–į–≤–Ľ–Ķ–Ĺ–ł—Ź –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ—Ź—é—ā CO –ł H2 —Ā –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –į–Ľ—Ć–ī–Ķ–≥–ł–ī–ĺ–≤[26]:

–ź–Ĺ–į–Ľ–ĺ–≥–ł—á–Ĺ–ĺ –Ņ—Ä–ĺ—ā–Ķ–ļ–į–Ķ—ā —Ä–Ķ–į–ļ—Ü–ł—Ź CO –ł H2O —Ā –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ļ–į—Ä–Ī–ĺ–Ĺ–ĺ–≤—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā[26] :

–ē—Ā–Ľ–ł –≤–ľ–Ķ—Ā—ā–ĺ –≤–ĺ–ī—č –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į—ā—Ć —Ā–Ņ–ł—Ä—ā, –ļ–ĺ–Ĺ–Ķ—á–Ĺ—č–ľ –Ņ—Ä–ĺ–ī—É–ļ—ā–ĺ–ľ —Ä–Ķ–į–ļ—Ü–ł–ł –Ī—É–ī–Ķ—ā —Ā–Ľ–ĺ–∂–Ĺ—č–Ļ —ć—Ą–ł—Ä[26] :

–†–Ķ–į–ļ—Ü–ł–ł –Ņ–ĺ–Ľ–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł–ł
–ü–ĺ–Ľ–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ľ–ĺ–∂–Ķ—ā –Ņ—Ä–ĺ—ā–Ķ–ļ–į—ā—Ć –ļ–į–ļ –Ņ–ĺ —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ–ĺ—Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–ľ—É, —ā–į–ļ –ł –ļ–į—ā–ł–ĺ–Ĺ–Ĺ–ĺ-–į–Ĺ–ł–ĺ–Ĺ–Ĺ–ĺ–ľ—É –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—É.
–ü–ĺ –Ņ–Ķ—Ä–≤–ĺ–ľ—É –ľ–Ķ—ā–ĺ–ī—É –Ņ–ĺ–Ľ—É—á–į—é—ā –Ņ–ĺ–Ľ–ł—ć—ā–ł–Ľ–Ķ–Ĺ –≤—č—Ā–ĺ–ļ–ĺ–≥–ĺ –ī–į–≤–Ľ–Ķ–Ĺ–ł—Ź:
![{\displaystyle {\mathsf {n\ CH_{2}\!\!=\!\!CH_{2}}}\rightarrow {\mathsf {-[-\!CH_{2}\!\!-\!\!CH_{2}\!-]_{n}-}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1e97cdfc9a0a959098c6ee2d45a5bc394ba41aa9)
–ö–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–ĺ–ľ —Ä–Ķ–į–ļ—Ü–ł–ł –≤—č—Ā—ā—É–Ņ–į—é—ā –Ņ–Ķ—Ä–ĺ–ļ—Ā–ł–ī—č.
–í—ā–ĺ—Ä–ĺ–Ļ –ľ–Ķ—ā–ĺ–ī –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ –≤ –ļ–į—á–Ķ—Ā—ā–≤–Ķ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–ĺ–≤ –ļ–ł—Ā–Ľ–ĺ—ā (–ļ–į—ā–ł–ĺ–Ĺ–Ĺ–į—Ź –Ņ–ĺ–Ľ–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł—Ź), –ľ–Ķ—ā–į–Ľ–Ľ–ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ (–ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä—č –¶–ł–≥–Ľ–Ķ—Ä–į-–Ě–į—ā—ā–į, –į–Ĺ–ł–ĺ–Ĺ–Ĺ–į—Ź –Ņ–ĺ–Ľ–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł—Ź). –ü—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤–ĺ–ľ –ľ–Ķ—ā–ĺ–ī–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā—Ć –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź —Ā—ā–Ķ—Ä–Ķ–ĺ—Ā–Ķ–Ľ–Ķ–ļ—ā–ł–≤–Ĺ—č—Ö –Ņ–ĺ–Ľ–ł–ľ–Ķ—Ä–ĺ–≤.
–†–Ķ–į–ļ—Ü–ł–ł —Ā–≤–ĺ–Ī–ĺ–ī–Ĺ–ĺ—Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź
–ú–Ķ—ā–į—ā–Ķ–∑–ł—Ā –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–í–Ņ–Ķ—Ä–≤—č–Ķ –ī–į–Ĺ–Ĺ—č–Ļ —ā–ł–Ņ —Ä–Ķ–į–ļ—Ü–ł–Ļ –Ī—č–Ľ –ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ –≤ —Ā–Ķ—Ä–Ķ–ī–ł–Ĺ–Ķ –Ņ—Ä–ĺ—ą–Ľ–ĺ–≥–ĺ –≤–Ķ–ļ–į –Ņ—Ä–ł –ł–∑—É—á–Ķ–Ĺ–ł–ł –Ņ–ĺ–Ľ–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł–ł —ć—ā–ł–Ľ–Ķ–Ĺ–į, –į –≤ –∑–į—ā–Ķ–ľ –Ī—č–Ľ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ –≤ 1966 –≥–ĺ–ī—É –ī–Ľ—Ź –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ —Ā–ł–Ĺ—ā–Ķ–∑–į –Ī—É—ā–Ķ–Ĺ–į-2.
–í 1967 –≥–ĺ–ī—É –Ě. –ö–į–Ľ—Ć–ī–Ķ—Ä–ĺ–Ĺ, –•. –ģ –ß–Ķ–Ĺ –ł –ö. –í. –°–ļ–ĺ—ā—ā –ĺ–Ņ–ł—Ā–į–Ľ–ł –ľ–Ķ—ā–į—ā–Ķ–∑–ł—Ā –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ (–≤ —Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ĺ–Ļ –Ľ–ł—ā–Ķ—Ä–į—ā—É—Ä–Ķ —á–į—Ā—ā–ĺ —É–Ņ–ĺ—ā—Ä–Ķ–Ī–Ľ—Ź–Ķ—ā—Ā—Ź —ā–Ķ—Ä–ľ–ł–Ĺ —Ä–Ķ–į–ļ—Ü–ł—Ź –ī–ł—Ā–ľ—É—ā–į—Ü–ł–ł –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤, –ł–Ĺ–į—á–Ķ –≥–ĺ–≤–ĺ—Ä—Ź ‚ÄĒ —Ä–Ķ–į–ļ—Ü–ł—é –ĺ–Ī–ľ–Ķ–Ĺ–į –į—ā–ĺ–ľ–į–ľ–ł –Ņ—Ä–ł —Ā–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł–ł –ĺ–Ī—Č–Ķ–Ļ —Ā—ā—Ä—É–ļ—ā—É—Ä—č –į–Ľ–ļ–Ķ–Ĺ–į –ł –Ķ–≥–ĺ –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł) –≤ —É—Ā–Ľ–ĺ–≤–ł—Ź—Ö –ļ–į—ā–į–Ľ–ł–∑–į —Ö–Ľ–ĺ—Ä–ł–ī–ĺ–ľ –≤–ĺ–Ľ—Ć—Ą—Ä–į–ľ–į (VI):

–†–Ķ–į–ļ—Ü–ł—Ź –ĺ–ļ–į–∑–į–Ľ–į—Ā—Ć –Ĺ–į—Ā—ā–ĺ–Ľ—Ć–ļ–ĺ –≤–į–∂–Ĺ–ĺ–Ļ –≤ –ĺ–Ī–Ľ–į—Ā—ā–ł –Ņ—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ł–≤–Ĺ–ĺ–Ļ —Ö–ł–ľ–ł–ł, —á—ā–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ć—Ā–ļ–į—Ź –≥—Ä—É–Ņ–Ņ–į –†–ĺ–Ī–Ķ—Ä—ā–į –ď—Ä–į–Ī–Ī—Ā–į, —Ä–į–∑—Ä–į–Ī–ĺ—ā–į–≤—ą–į—Ź –Ĺ–ĺ–≤—č–Ļ –ļ–Ľ–į—Ā—Ā –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–ĺ–≤ (–į–Ľ–ļ–ł–Ľ–ł–ī–Ķ–Ĺ–ĺ–≤—č–Ķ –ļ–ĺ–ľ–Ņ–Ľ–Ķ–ļ—Ā—č —Ä—É—ā–Ķ–Ĺ–ł—Ź) –ľ–Ķ—ā–į—ā–Ķ–∑–ł—Ā–į –ĺ–Ľ–Ķ—Ą–ł–Ĺ–ĺ–≤, –Ņ–ĺ–Ľ—É—á–ł–Ľ–į –≤ 2005 –≥–ĺ–ī—É –Ě–ĺ–Ī–Ķ–Ľ–Ķ–≤—Ā–ļ—É—é –Ņ—Ä–Ķ–ľ–ł—é –≤ –ĺ–Ī–Ľ–į—Ā—ā–ł —Ö–ł–ľ–ł–ł[27]. –≠—ā—É –Ņ—Ä–Ķ–ľ–ł—é —ā–į–ļ–∂–Ķ –Ņ–ĺ–Ľ—É—á–ł–Ľ–ł —Ą—Ä–į–Ĺ—Ü—É–∑ –ė–≤ –®–ĺ–≤–Ķ–Ĺ –≤ 1971 –≥–ĺ–ī—É, –Ņ—Ä–Ķ–ī–Ľ–ĺ–∂–ł–≤—ą–ł–Ļ –ļ–į—Ä–Ī–Ķ–Ĺ–ĺ–≤—É—é —ā–Ķ–ĺ—Ä–ł—é –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–į —Ä–Ķ–į–ļ—Ü–ł–ł –ľ–Ķ—ā–į—ā–Ķ–∑–ł—Ā–į[28], –ł –į–ľ–Ķ—Ä–ł–ļ–į–Ĺ–Ķ—Ü –†–ł—á–į—Ä–ī –®—Ä–ĺ–ļ, —Ā–ĺ–∑–ī–į–≤—ą–ł–Ļ –≤ 1990 –≥–ĺ–ī—É –Ņ–Ķ—Ä–≤—č–Ļ –ľ–Ķ—ā–į–Ľ–Ľ–ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ļ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä –ľ–Ķ—ā–į—ā–Ķ–∑–ł—Ā–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤[29].
–í 2008 –≥–ĺ–ī—É –Ņ–ĺ–Ľ—Ć—Ā–ļ–ł–Ķ —Ö–ł–ľ–ł–ļ–ł –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ–ł —Ä–Ķ–į–ļ—Ü–ł—é –ľ–Ķ—ā–į—ā–Ķ–∑–ł—Ā–į –≤ –≤–ĺ–ī–Ĺ–ĺ–ľ —Ä–į—Ā—ā–≤–ĺ—Ä–Ķ —Ā –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ļ–ĺ–ľ–ľ–Ķ—Ä—á–Ķ—Ā–ļ–ł –ī–ĺ—Ā—ā—É–Ņ–Ĺ–ĺ–≥–ĺ —Ä—É—ā–Ķ–Ĺ–ł–Ķ–≤–ĺ–≥–ĺ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į[30].
–Ę–Ķ—Ö–Ĺ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ –į—Ā–Ņ–Ķ–ļ—ā—č –ľ–Ķ—ā–į—ā–Ķ–∑–ł—Ā–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ —Ä–į—Ā—Ā–ľ–ĺ—ā—Ä–Ķ–Ĺ—č –≤ —Ā—ā–į—ā—Ć–Ķ: –ú–Ķ—ā–į—ā–Ķ–∑–ł—Ā –ĺ–Ľ–Ķ—Ą–ł–Ĺ–ĺ–≤: —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ—č–Ļ –Ņ—É—ā—Ć –ļ –Ņ–ĺ–Ľ–ł–Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ—É.
–ú–Ķ—ā–ĺ–ī—č –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–ě—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ—č–ľ –ľ–Ķ—ā–ĺ–ī–ĺ–ľ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ļ–į—ā–į–Ľ–ł—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –ł –≤—č—Ā–ĺ–ļ–ĺ—ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–Ĺ—č–Ļ –ļ—Ä–Ķ–ļ–ł–Ĺ–≥ —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī–ĺ–≤ –Ĺ–Ķ—Ą—ā–ł –ł –Ņ—Ä–ł—Ä–ĺ–ī–Ĺ–ĺ–≥–ĺ –≥–į–∑–į. –Ē–Ľ—Ź –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į –Ĺ–ł–∑—ą–ł—Ö –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā —ā–į–ļ–∂–Ķ —Ä–Ķ–į–ļ—Ü–ł—é –ī–Ķ–≥–ł–ī—Ä–į—ā–į—Ü–ł–ł —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—Č–ł—Ö —Ā–Ņ–ł—Ä—ā–ĺ–≤.
–í –Ľ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–Ĺ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ –ĺ–Ī—č—á–Ĺ–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—é—ā –ľ–Ķ—ā–ĺ–ī –ī–Ķ–≥–ł–ī—Ä–į—ā–į—Ü–ł–ł —Ā–Ņ–ł—Ä—ā–ĺ–≤ –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł —Ā–ł–Ľ—Ć–Ĺ—č—Ö –ľ–ł–Ĺ–Ķ—Ä–į–Ľ—Ć–Ĺ—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā[2], –ī–Ķ–≥–ł–ī—Ä–ĺ–≥–į–Ľ–ĺ–≥–Ķ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ł –ī–Ķ–≥–į–Ľ–ĺ–≥–Ķ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—Č–ł—Ö –≥–į–Ľ–ĺ–≥–Ķ–Ĺ–Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č—Ö; —Ā–ł–Ĺ—ā–Ķ–∑—č –ď–ĺ—Ą–ľ–į–Ĺ–į, –ß—É–≥–į–Ķ–≤–į, –í–ł—ā—ā–ł–≥–į –ł –ö–ĺ—É–Ņ–į[31].
–ü–ĺ–ī—Ä–ĺ–Ī–Ĺ–Ķ–Ķ ‚ÄĒ —Ā–ľ. —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—Č–ł–Ķ —Ä–į–∑–ī–Ķ–Ľ—č –Ĺ–ł–∂–Ķ.
–Ē–Ķ–≥–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į–Ľ–ļ–į–Ĺ–ĺ–≤
–≠—ā–ĺ –ĺ–ī–ł–Ĺ –ł–∑ –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ—č—Ö —Ā–Ņ–ĺ—Ā–ĺ–Ī–ĺ–≤ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤[32][33]. –Ę–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–į: 350‚ÄĒ450 ¬įC, –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä ‚ÄĒ Cr2O3. –Ę–į–ļ–∂–Ķ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā—Ā—Ź –į–Ľ—é–ľ–ĺ–ľ–ĺ–Ľ–ł–Ī–ī–Ķ–Ĺ–ĺ–≤—č–Ķ –ł –į–Ľ—é–ľ–ĺ–Ņ–Ľ–į—ā–ł–Ĺ–ĺ–≤—č–Ķ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä—č[34]. –Ē–Ľ—Ź –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź —ā—Ä–į–Ĺ—Ā-–į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā MOH/EtOH, –ī–Ľ—Ź —Ü–ł—Ā-–Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č—Ö NaNH2/NH3


–Ē–Ķ–≥–ł–ī—Ä–ĺ–≥–į–Ľ–ĺ–≥–Ķ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ł –ī–Ķ–≥–į–Ľ–ĺ–≥–Ķ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į–Ľ–ļ–į–Ĺ–ĺ–≤
–ě—ā—Č–Ķ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ –≥–į–Ľ–ĺ–≥–Ķ–Ĺ–ĺ–≤ —É –ī–ł–≥–į–Ľ–ĺ–≥–Ķ–Ĺ–į–Ľ–ļ–į–Ĺ–ĺ–≤ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł —Ü–ł–Ĺ–ļ–į[35]:

–Ē–Ķ–≥–ł–ī—Ä–ĺ–≥–į–Ľ–ĺ–≥–Ķ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –Ņ—Ä–ĺ–≤–ĺ–ī—Ź—ā –Ņ—Ä–ł –Ĺ–į–≥—Ä–Ķ–≤–į–Ĺ–ł–ł –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ —Ā–Ņ–ł—Ä—ā–ĺ–≤—č–ľ–ł —Ä–į—Ā—ā–≤–ĺ—Ä–į–ľ–ł —Č–Ķ–Ľ–ĺ—á–Ķ–Ļ[36]:

–ü—Ä–ł –ĺ—ā—Č–Ķ–Ņ–Ľ–Ķ–Ĺ–ł–ł –≥–į–Ľ–ĺ–≥–Ķ–Ĺ–≤–ĺ–ī–ĺ—Ä–ĺ–ī–į –ĺ–Ī—Ä–į–∑—É–Ķ—ā—Ā—Ź —Ā–ľ–Ķ—Ā—Ć –ł–∑–ĺ–ľ–Ķ—Ä–ĺ–≤, –Ņ—Ä–Ķ–ĺ–Ī–Ľ–į–ī–į—é—Č–ł–Ļ –ł–∑ –ļ–ĺ—ā–ĺ—Ä—č—Ö –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ—Ä–į–≤–ł–Ľ–ĺ–ľ –ó–į–Ļ—Ü–Ķ–≤–į: –ĺ—ā—Č–Ķ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ĺ—ā–ĺ–Ĺ–į –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –ĺ—ā –ľ–Ķ–Ĺ–Ķ–Ķ –≥–ł–ī—Ä–ĺ–≥–Ķ–Ĺ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –į—ā–ĺ–ľ–į —É–≥–Ľ–Ķ—Ä–ĺ–ī–į.
–Ē–Ķ–≥–ł–ī—Ä–į—ā–į—Ü–ł—Ź —Ā–Ņ–ł—Ä—ā–ĺ–≤
–Ē–Ķ–≥–ł–ī—Ä–į—ā–į—Ü–ł—é —Ā–Ņ–ł—Ä—ā–ĺ–≤ –≤–Ķ–ī—É—ā –Ņ—Ä–ł –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ–ľ–Ņ–Ķ—Ä–į—ā—É—Ä–Ķ –Ī–į–Ĺ–ł –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł —Ā–ł–Ľ—Ć–Ĺ—č—Ö –ľ–ł–Ĺ–Ķ—Ä–į–Ľ—Ć–Ĺ—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā[35]:


–í —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ –į–Ľ–ļ–Ķ–Ĺ—č –ł–∑ –≤—ā–ĺ—Ä–ł—á–Ĺ—č—Ö –ł —ā—Ä–Ķ—ā–ł—á–Ĺ—č—Ö —Ā–Ņ–ł—Ä—ā–ĺ–≤ —ā–į–ļ–∂–Ķ –Ņ–ĺ–Ľ—É—á–į—é—ā —Ā –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ī–Ķ–≥–ł–ī—Ä–į—ā–ł—Ä—É—é—Č–Ķ–≥–ĺ —Ä–Ķ–į–≥–Ķ–Ĺ—ā–į ‚ÄĒ —Ä–Ķ–į–≥–Ķ–Ĺ—ā–į –Ď—Ď—Ä–ī–∂–Ķ—Ā—Ā–į[20]:

–ď–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į–Ľ–ļ–ł–Ĺ–ĺ–≤
–ß–į—Ā—ā–ł—á–Ĺ–ĺ–Ķ –≥–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į–Ľ–ļ–ł–Ĺ–ĺ–≤ —ā—Ä–Ķ–Ī—É–Ķ—ā —Ā–Ņ–Ķ—Ü–ł–į–Ľ—Ć–Ĺ—č—Ö —É—Ā–Ľ–ĺ–≤–ł–Ļ –ł –Ĺ–į–Ľ–ł—á–ł–Ķ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į (–Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä, –ī–Ķ–∑–į–ļ—ā–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ–į–Ľ–Ľ–į–ī–ł—Ź ‚ÄĒ –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–į –õ–ł–Ĺ–ī–Ľ–į—Ä–į)[35]:
 (—Ü–ł—Ā-–ł–∑–ĺ–ľ–Ķ—Ä)
(—Ü–ł—Ā-–ł–∑–ĺ–ľ–Ķ—Ä)
 (—ā—Ä–į–Ĺ—Ā-–ł–∑–ĺ–ľ–Ķ—Ä)
(—ā—Ä–į–Ĺ—Ā-–ł–∑–ĺ–ľ–Ķ—Ä) 
–†–Ķ–į–ļ—Ü–ł—Ź –í–ł—ā—ā–ł–≥–į
–†–Ķ–į–ļ—Ü–ł—Ź –í–ł—ā—ā–ł–≥–į ‚ÄĒ —Ā—ā–Ķ—Ä–Ķ–ĺ—Ā–Ķ–Ľ–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ —Ā–ł–Ĺ—ā–Ķ–∑ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –ļ–į—Ä–Ī–ĺ–Ĺ–ł–Ľ—Ć–Ĺ—č—Ö —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ –ł –į–Ľ–ļ–ł–Ľ–ł–ī–Ķ–Ĺ—Ą–ĺ—Ā—Ą–ĺ—Ä–į–Ĺ–ĺ–≤ (–ł–Ľ–ł–ī–ĺ–≤ —Ą–ĺ—Ā—Ą–ĺ–Ĺ–ł–Ķ–≤—č—Ö —Ā–ĺ–Ľ–Ķ–Ļ)[37]:
![{\displaystyle {\mathsf {(C_{6}H_{5})_{3}P+CH_{3}Br}}\rightarrow {\mathsf {[(C_{6}H_{5})_{3}P\!\!-\!\!CH_{3}]Br}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/73a40add539caa3e89155b37347220e6410e653c)
![{\displaystyle {\mathsf {[(C_{6}H_{5})_{3}P\!\!-\!\!CH_{3}]Br+C_{6}H_{5}Li}}\rightarrow {\mathsf {(C_{6}H_{5})_{3}P\!\!-\!\!CH_{2}\!\!:+}}{\mathsf {C_{6}H_{6}+LiBr}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ce8a7eb853879473cc1c355a6a154aaf1711f5ad)

–Ē–Ľ—Ź –Ņ—Ä–Ķ–≤—Ä–į—Č–Ķ–Ĺ–ł—Ź —Ā–ĺ–Ľ–Ķ–Ļ —Ą–ĺ—Ā—Ą–ĺ–Ĺ–ł—Ź –≤ –ł–Ľ–ł–ī—č –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā—Ā—Ź –Ī—É—ā–ł–Ľ–Ľ–ł—ā–ł–Ļ, –≥–ł–ī—Ä–ł–ī, –į–ľ–ł–ī –ł–Ľ–ł –į–Ľ–ļ–ĺ–≥–ĺ–Ľ—Ź—ā –Ĺ–į—ā—Ä–ł—Ź, –į —ā–į–ļ–∂–Ķ –Ĺ–Ķ–ļ–ĺ—ā–ĺ—Ä—č–Ķ –ī—Ä—É–≥–ł–Ķ —Ā–ł–Ľ—Ć–Ĺ—č–Ķ –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł—Ź.
–í —Ä–Ķ–į–ļ—Ü–ł—é –ľ–ĺ–≥—É—ā –≤—Ā—ā—É–Ņ–į—ā—Ć —Ā–į–ľ—č–Ķ —Ä–į–∑–Ľ–ł—á–Ĺ—č–Ķ –ļ–į—Ä–Ī–ĺ–Ĺ–ł–Ľ—Ć–Ĺ—č–Ķ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź, —Ā—Ä–Ķ–ī–ł –ļ–ĺ—ā–ĺ—Ä—č—Ö –į—Ä–ĺ–ľ–į—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –ł –į–Ľ–ł—Ą–į—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –į–Ľ—Ć–ī–Ķ–≥–ł–ī—č –ł –ļ–Ķ—ā–ĺ–Ĺ—č, –≤ —ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ —Ā–ĺ–ī–Ķ—Ä–∂–į—Č–ł–Ķ –ī–≤–ĺ–Ļ–Ĺ—č–Ķ –ł —ā—Ä–ĺ–Ļ–Ĺ—č–Ķ —Ā–≤—Ź–∑–ł –ł —Ä–į–∑–Ľ–ł—á–Ĺ—č–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č–Ķ –≥—Ä—É–Ņ–Ņ—č.
–í –Ľ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–Ĺ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ —á–į—Ā—ā–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā –Ī–ĺ–Ľ–Ķ–Ķ —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ—É—é –ľ–ĺ–ī–ł—Ą–ł–ļ–į—Ü–ł—é (1959 –≥–ĺ–ī) —Ä–Ķ–į–ļ—Ü–ł–ł –í–ł—ā—ā–ł–≥–į ‚ÄĒ —Ä–Ķ–į–ļ—Ü–ł—é –•–ĺ—Ä–Ĺ–Ķ—Ä–į-–£–ĺ–ī—Ā–≤–ĺ—Ä—ā–į-–≠–ľ–ľ–ĺ–Ĺ—Ā–į[38]:
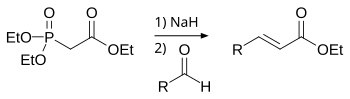
–ü—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź —Ą–ĺ—Ā—Ą–ĺ–Ĺ–į—ā–ĺ–≤ –∑–į–ļ–Ľ—é—á–į–Ķ—ā—Ā—Ź –≤ —ā–ĺ–ľ, —á—ā–ĺ –ĺ–Ī—Ä–į–∑—É—é—Č–ł–Ķ—Ā—Ź –≤ —Ö–ĺ–ī–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł —Ą–ĺ—Ā—Ą–į—ā—č –Ľ–Ķ–≥–ļ–ĺ –ĺ—ā–ľ—č–≤–į—é—ā—Ā—Ź –≤–ĺ–ī–ĺ–Ļ. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, —Ä–Ķ–į–ļ—Ü–ł—Ź –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź–Ķ—ā –ł–∑–Ī–ł—Ä–į—ā—Ć –ĺ–Ņ—ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł–Ķ —ć–Ľ–ł–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź, –Ņ–ĺ–Ľ—É—á–į—Ź –Ĺ–į –≤—č—Ö–ĺ–ī–Ķ —ā—Ä–į–Ĺ—Ā- (—ā–Ķ—Ä–ľ–ĺ–ī–ł–Ĺ–į–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ć) –ł–Ľ–ł —Ü–ł—Ā-–ł–∑–ĺ–ľ–Ķ—Ä—č (–ļ–ł–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ć)[20].
–†–Ķ–į–ļ—Ü–ł—Ź –ö–Ĺ—Ď–≤–Ķ–Ĺ–į–≥–Ķ–Ľ—Ź
–†–Ķ–į–ļ—Ü–ł—Ź –ö–Ĺ—Ď–≤–Ķ–Ĺ–į–≥–Ķ–Ľ—Ź ‚ÄĒ –ļ–ĺ–Ĺ–ī–Ķ–Ĺ—Ā–į—Ü–ł—Ź –į–Ľ—Ć–ī–Ķ–≥–ł–ī–ĺ–≤ –ł–Ľ–ł –ļ–Ķ—ā–ĺ–Ĺ–ĺ–≤ —Ā —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź–ľ–ł, —Ā–ĺ–ī–Ķ—Ä–∂–į—Č–ł–ľ–ł –į–ļ—ā–ł–≤–Ĺ—É—é CH2-–≥—Ä—É–Ņ–Ņ—É[20]:

–†–Ķ–į–ļ—Ü–ł—Ź –ł–ľ–Ķ–Ķ—ā –ĺ—á–Ķ–Ĺ—Ć —ą–ł—Ä–ĺ–ļ–ł–Ļ –ī–ł–į–Ņ–į–∑–ĺ–Ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź, –Ņ—Ä–ł —ć—ā–ĺ–ľ –Ņ–ĺ–ľ–ł–ľ–ĺ —ć—Ą–ł—Ä–ĺ–≤ –ľ–į–Ľ–ĺ–Ĺ–ĺ–≤–ĺ–Ļ –ļ–ł—Ā–Ľ–ĺ—ā—č, –≤ —Ä–Ķ–į–ļ—Ü–ł—é –ľ–ĺ–≥—É—ā –≤—Ā—ā—É–Ņ–į—ā—Ć –ł –ī—Ä—É–≥–ł–Ķ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź, –Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä: CH3CN, CH3NO2, LiCH2COOC2H5 –ł –Ņ—Ä.[14].
–†–Ķ–į–ļ—Ü–ł—Ź –ß—É–≥–į–Ķ–≤–į
–†–Ķ–į–ļ—Ü–ł—Ź –ß—É–≥–į–Ķ–≤–į ‚ÄĒ –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ —Ā–Ņ–ł—Ä—ā–ĺ–≤ —Ā CS2 –ł NaOH —Ā –Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–ł–ľ –ľ–Ķ—ā–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ł –ī–į–Ľ—Ć–Ĺ–Ķ–Ļ—ą–ł–ľ –Ņ–ł—Ä–ĺ–Ľ–ł–∑–ĺ–ľ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–≤—ą–ł—Ö—Ā—Ź S-–ľ–Ķ—ā–ł–Ľ–ļ—Ā–į–Ĺ—ā–ĺ–≥–Ķ–Ĺ–į—ā–ĺ–≤[39]:


–†–Ķ–į–ļ—Ü–ł—Ź –ď–ĺ—Ą–ľ–į–Ĺ–į
–ė—Ā—á–Ķ—Ä–Ņ—č–≤–į—é—Č–Ķ–Ķ –ľ–Ķ—ā–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –Ņ–ĺ –ď–ĺ—Ą–ľ–į–Ĺ—É ‚ÄĒ —Ä–į–∑–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ —á–Ķ—ā–≤–Ķ—Ä—ā–ł—á–Ĺ—č—Ö –į–ľ–ľ–ĺ–Ĺ–ł–Ķ–≤—č—Ö –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–Ļ –Ĺ–į –į–Ľ–ļ–Ķ–Ĺ, —ā—Ä–Ķ—ā–ł—á–Ĺ—č–Ļ –į–ľ–ł–Ĺ –ł –≤–ĺ–ī—É[40]:

–Ě–į –Ņ–Ķ—Ä–≤–ĺ–Ļ —Ā—ā–į–ī–ł–ł —Ä–Ķ–į–ļ—Ü–ł–ł –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –ľ–Ķ—ā–ł–Ľ–ł–ĺ–ī–ł–ī–į –į–ľ–ł–Ĺ –Ņ—Ä–Ķ–≤—Ä–į—Č–į—é—ā –≤ —á–Ķ—ā–≤–Ķ—Ä—ā–ł—á–Ĺ—č–Ļ –į–ľ–ľ–ĺ–Ĺ–ł–Ļ–ł–ĺ–ī–ł–ī, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ī–į–Ľ–Ķ–Ķ –Ņ–Ķ—Ä–Ķ–≤–ĺ–ī—Ź—ā –≤ –≥–ł–ī—Ä–ĺ–ļ—Ā–ł–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –ĺ–ļ—Ā–ł–ī–į —Ā–Ķ—Ä–Ķ–Ī—Ä–į, –Ĺ–į–ļ–ĺ–Ĺ–Ķ—Ü, –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ļ —ć—ā–į–Ņ ‚ÄĒ —Ä–į–∑–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ ‚ÄĒ –≤–Ķ–ī—É—ā –Ņ—Ä–ł 100‚ÄĒ200 ¬įC, —á–į—Ā—ā–ĺ –Ņ—Ä–ł –Ņ–ĺ–Ĺ–ł–∂–Ķ–Ĺ–Ĺ–ĺ–ľ –ī–į–≤–Ľ–Ķ–Ĺ–ł–ł[41].
–≠–Ľ–ł–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –Ņ–ĺ –ď–ĺ—Ą–ľ–į–Ĺ—É –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—é –Ĺ–į–ł–ľ–Ķ–Ĺ–Ķ–Ķ –∑–į–ľ–Ķ—Č—Ď–Ĺ–Ĺ—č—Ö –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ (–Ņ—Ä–ĺ—ā–ł–≤ –Ņ—Ä–į–≤–ł–Ľ–į –ó–į–Ļ—Ü–Ķ–≤–į).
–ú–Ķ—ā–ĺ–ī –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ—ā—Ā—Ź, –≤ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–ľ, –ī–Ľ—Ź –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –Ĺ–Ķ–ļ–ĺ—ā–ĺ—Ä—č—Ö —Ü–ł–ļ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ł –≤ —Ö–ł–ľ–ł–ł –į–Ľ–ļ–į–Ľ–ĺ–ł–ī–ĺ–≤[41].
–†–Ķ–į–ļ—Ü–ł—Ź –ö–ĺ—É–Ņ–į
–†–Ķ–į–ļ—Ü–ł—Ź –ö–ĺ—É–Ņ–į[–į–Ĺ–≥–Ľ.] ‚ÄĒ —Ä–į–∑–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ N-–ĺ–ļ–ł—Ā–Ķ–Ļ —ā—Ä–Ķ—ā–ł—á–Ĺ—č—Ö –į–ľ–ł–Ĺ–ĺ–≤[41]:

–ü—Ä–ĺ—á–ł–Ķ –ľ–Ķ—ā–ĺ–ī—č —Ā–ł–Ĺ—ā–Ķ–∑–į
–†–Ķ–į–ļ—Ü–ł—Ź –Ď—É—Ä–ī–į
–†–Ķ–į–ļ—Ü–ł—Ź –Ď—É—Ä–ī–į ‚ÄĒ —ć–Ľ–ł–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –Ī—Ä–ĺ–ľ–į –ł —ć—ā–ĺ–ļ—Ā–ł–≥—Ä—É–Ņ–Ņ—č –ł–∑ –Ī—Ä–ĺ–ľ–į–Ľ–ļ–ł–Ľ—ć—ā–ł–Ľ–ĺ–≤—č—Ö —ć—Ą–ł—Ä–ĺ–≤ –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ —Ü–ł–Ĺ–ļ–ĺ–≤–ĺ–Ļ –Ņ—č–Ľ–ł[42]:

–°–ł–Ĺ—ā–Ķ–∑ –ł–∑ —ā–ĺ–∑–ł–Ľ–≥–ł–ī—Ä–į–∑–ĺ–Ĺ–ĺ–≤
–ź–Ľ–ļ–Ķ–Ĺ—č –ľ–ĺ–∂–Ĺ–ĺ –Ņ–ĺ–Ľ—É—á–ł—ā—Ć —Ä–į–∑–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ–ľ —ā–ĺ–∑–ł–Ľ–≥–ł–ī—Ä–į–∑–ĺ–Ĺ–ĺ–≤ –Ņ–ĺ–ī –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ–ľ –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–Ļ (–†–Ķ–į–ļ—Ü–ł—Ź –Ď—ć–ľ—Ą–ĺ—Ä–ī–į ‚ÄĒ –°—ā–ł–≤–Ķ–Ĺ—Ā–į[–į–Ĺ–≥–Ľ.] –ł –†–Ķ–į–ļ—Ü–ł—Ź –®–į–Ņ–ł—Ä–ĺ[–į–Ĺ–≥–Ľ.])[43]:

–†–Ķ–į–ļ—Ü–ł—Ź –Ď—ć–ľ—Ą–ĺ—Ä–ī–į ‚ÄĒ –°—ā–ł–≤–Ķ–Ĺ—Ā–į –ł –†–Ķ–į–ļ—Ü–ł—Ź –®–į–Ņ–ł—Ä–ĺ –Ņ—Ä–ĺ—ā–Ķ–ļ–į—é—ā –Ņ–ĺ –ĺ–ī–ł–Ĺ–į–ļ–ĺ–≤–ĺ–ľ—É –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—É.
–í –Ņ–Ķ—Ä–≤–ĺ–ľ —Ā–Ľ—É—á–į–Ķ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā—Ā—Ź –Ĺ–į—ā—Ä–ł–Ļ, –ľ–Ķ—ā–ł–Ľ–į—ā –Ĺ–į—ā—Ä–ł—Ź, –≥–ł–ī—Ä–ł–ī—č –Ľ–ł—ā–ł—Ź –ł–Ľ–ł –Ĺ–į—ā—Ä–ł—Ź, –į–ľ–ł–ī –Ĺ–į—ā—Ä–ł—Ź –ł —ā. –Ņ. –í–ĺ –≤—ā–ĺ—Ä–ĺ–ľ: –į–Ľ–Ľ–ļ–ł–Ľ–Ľ–ł—ā–ł–Ļ –ł —Ä–Ķ–į–ļ—ā–ł–≤—č –ď—Ä–ł–Ĺ—Ć—Ź—Ä–į.
–í —Ä–Ķ–į–ļ—Ü–ł—Ź –Ď—ć–ľ—Ą–ĺ—Ä–ī–į ‚ÄĒ –°—ā–ł–≤–Ķ–Ĺ—Ā–į –ĺ–Ī—Ä–į–∑—É—é—ā—Ā—Ź –Ī–ĺ–Ľ–Ķ–Ķ –∑–į–ľ–Ķ—Č—Ď–Ĺ–Ĺ—č–Ķ, –į –≤ —Ä–Ķ–į–ļ—Ü–ł—Ź –®–į–Ņ–ł—Ä–ĺ ‚ÄĒ –Ĺ–į–ł–ľ–Ķ–Ĺ–Ķ–Ķ –∑–į–ľ–Ķ—Č—Ď–Ĺ–Ĺ—č–Ķ –į–Ľ–ļ–Ķ–Ĺ—č[44].
–†–Ķ–į–ļ—Ü–ł—Ź –ü–Ķ—Ä–ļ–ł–Ĺ–į
–†–Ķ–į–ļ—Ü–ł—Ź –ü–Ķ—Ä–ļ–ł–Ĺ–į ‚ÄĒ –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ –į—Ä–ĺ–ľ–į—ā–ł—á–Ķ—Ā–ļ–ł—Ö –į–Ľ—Ć–ī–Ķ–≥–ł–ī–ĺ–≤ —Ā –į–Ĺ–≥–ł–ī—Ä–ł–ī–į–ľ–ł –ļ–į—Ä–Ī–ĺ–Ĺ–ĺ–≤—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā –≤ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ł–ł –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä–ĺ–≤ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–≥–ĺ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–į (—Č–Ķ–Ľ–ĺ—á–Ĺ—č—Ö —Ā–ĺ–Ľ–Ķ–Ļ –ļ–į—Ä–Ī–ĺ–Ĺ–ĺ–≤—č—Ö –ļ–ł—Ā–Ľ–ĺ—ā, —ā—Ä–Ķ—ā–ł—á–Ĺ—č—Ö –į–ľ–ł–Ĺ–ĺ–≤ –ł —ā. –Ņ.)[45]:

–ü–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–ł–ľ –ī–Ķ–ļ–į—Ä–Ī–ĺ–ļ—Ā–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ĺ–Ī—Ä–į–∑—É—é—Č–Ķ–Ļ—Ā—Ź –ļ–ł—Ā–Ľ–ĺ—ā—č –ľ–ĺ–∂–Ĺ–ĺ –Ņ–ĺ–Ľ—É—á–ł—ā—Ć —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—Č–ł–Ļ –į–Ľ–ļ–Ķ–Ĺ.
–°–ł–Ĺ—ā–Ķ–∑ –ö–ĺ—Ä–ł ‚ÄĒ –í–ł–Ĺ—ā–Ķ—Ä–į

–ě–Ľ–Ķ—Ą–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –Ė—é–Ľ–ł–į ‚ÄĒ –õ–ł–∂–ĺ

–ė–ī–Ķ–Ĺ—ā–ł—Ą–ł–ļ–į—Ü–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—ā–ĺ–ī—č –ł–ī–Ķ–Ĺ—ā–ł—Ą–ł–ļ–į—Ü–ł–ł –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–ß–į—Ā—ā–ĺ –ī–Ľ—Ź –ł–ī–Ķ–Ĺ—ā–ł—Ą–ł–ļ–į—Ü–ł–ł –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā —Ä–Ķ–į–ļ—Ü–ł—é –í–į–≥–Ĺ–Ķ—Ä–į: –ĺ–Ī–Ķ—Ā—Ü–≤–Ķ—á–ł–≤–į–Ĺ–ł–Ķ —Ä–į—Ā—ā–≤–ĺ—Ä–į –Ņ–Ķ—Ä–ľ–į–Ĺ–≥–į–Ĺ–į—ā–į –ļ–į–Ľ–ł—Ź –≤ —Ā–Ľ–į–Ī–ĺ—Č–Ķ–Ľ–ĺ—á–Ĺ–ĺ–Ļ —Ā—Ä–Ķ–ī–Ķ (–ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ī–ĺ –≥–Ľ–ł–ļ–ĺ–Ľ–Ķ–Ļ).
–Ē—Ä—É–≥–ĺ–Ļ –≤–į—Ä–ł–į–Ĺ—ā ‚ÄĒ –ĺ–Ī–Ķ—Ā—Ü–≤–Ķ—á–ł–≤–į–Ĺ–ł–Ķ —Ä–į—Ā—ā–≤–ĺ—Ä–į –Ī—Ä–ĺ–ľ–į –≤ —á–Ķ—ā—č—Ä–Ķ—Ö—Ö–Ľ–ĺ—Ä–ł—Ā—ā–ĺ–ľ —É–≥–Ľ–Ķ—Ä–ĺ–ī–Ķ –Ņ—Ä–ł –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–ł –≤—č–ī–Ķ–Ľ–Ķ–Ĺ–ł—Ź –Ī—Ä–ĺ–ľ–ĺ–≤–ĺ–ī–ĺ—Ä–ĺ–ī–į (—Ä–Ķ–į–ļ—Ü–ł—Ź –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź)[46].
–≠—ā–ł —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—ā–ĺ–ī—č —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ĺ—á–Ķ–Ĺ—Ć –ĺ–Ī—Č–ł–ľ–ł, –Ĺ–Ķ —Ā–Ķ–Ľ–Ķ–ļ—ā–ł–≤–Ĺ—č–ľ–ł –ł –Ĺ–Ķ –ľ–ĺ–≥—É—ā –≥–į—Ä–į–Ĺ—ā–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–ł—ā—Ć –į–Ľ–ļ–Ķ–Ĺ—č.
–Ē–Ľ—Ź –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–Ķ–Ĺ–ł—Ź –Ĺ–į–Ľ–ł—á–ł—Ź –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ —Ā–≤—Ź–∑–ł –≤ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā –ľ–Ķ—ā–ĺ–ī—č —Ā–Ņ–Ķ–ļ—ā—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł–ł.
–ú–į—Ā—Ā-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—ā–ĺ–ī—č –į–Ĺ–į–Ľ–ł–∑–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–ú–į—Ā—Ā-—Ā–Ņ–Ķ–ļ—ā—Ä—č –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā –į–Ľ–ļ–į–Ĺ–į–ľ–ł —Ā–ĺ–ī–Ķ—Ä–∂–į—ā –Ī–ĺ–Ľ–Ķ–Ķ –ł–Ĺ—ā–Ķ–Ĺ—Ā–ł–≤–Ĺ—č–Ķ M+ –Ņ–ł–ļ–ł[47].
–°—É—Č–Ķ—Ā—ā–≤—É–Ķ—ā —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā-–ľ–Ķ—ā–ĺ–ī –ľ–į—Ā—Ā-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ–ľ–Ķ—ā—Ä–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ā—ā—Ä–ĺ–Ķ–Ĺ–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤, –∑–į–ļ–Ľ—é—á–į—é—Č–ł–Ļ—Ā—Ź –≤ –ł–∑—É—á–Ķ–Ĺ–ł–ł –ľ–į—Ā—Ā-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ–≤ —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—Č–ł—Ö –į–Ľ–ļ–į–Ĺ–ĺ–≤, –ĺ–Ī—Ä–į–∑—É—é—Č–ł—Ö—Ā—Ź –Ņ—Ä–ł –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł–ł –≥–į–∑–ĺ—Ą–į–∑–Ĺ–ĺ–≥–ĺ –≥–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –≤ —ā–ĺ–ļ–Ķ –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į (–ļ–į—ā. Pt, Pd) –≤ –ľ–ł–ļ—Ä–ĺ—Ä–Ķ–į–ļ—ā–ĺ—Ä–Ķ, —Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–Ĺ–ĺ–ľ –ľ–Ķ–∂–ī—É –≥–į–∑–ĺ–≤—č–ľ —Ö—Ä–ĺ–ľ–į—ā–ĺ–≥—Ä–į—Ą–ĺ–ľ –ł –ľ–į—Ā—Ā-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ–ľ–Ķ—ā—Ä–ĺ–ľ[48].
–£–§-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—ā–ĺ–ī—č –į–Ĺ–į–Ľ–ł–∑–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–ź–Ľ–ļ–Ķ–Ĺ—č —Ā –ł–∑–ĺ–Ľ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–ľ–ł –ī–≤–ĺ–Ļ–Ĺ—č–ľ–ł —Ā–≤—Ź–∑—Ź–ľ–ł –ł–ľ–Ķ—é—ā –ł–Ĺ—ā–Ķ–Ĺ—Ā–ł–≤–Ĺ—É—é (őĶ –ĺ—ā 6500 –ī–ĺ 12000) —ą–ł—Ä–ĺ–ļ—É—é –Ņ–ĺ–Ľ–ĺ—Ā—É –Ņ–ĺ–≥–Ľ–ĺ—Č–Ķ–Ĺ–ł—Ź, –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ—É—é –Ņ–Ķ—Ä–Ķ—Ö–ĺ–ī–ĺ–ľ ŌÄ‚ÜíŌÄ, –≤ –ĺ–Ī–Ľ–į—Ā—ā–ł 165‚ÄĒ200 –Ĺ–ľ. –Ě–į–Ľ–ł—á–ł–Ķ –į–Ľ–ļ–ł–Ľ—Ć–Ĺ—č—Ö –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–Ķ–Ļ —Ā–ľ–Ķ—Č–į–Ķ—ā —ć—ā—É –Ņ–ĺ–Ľ–ĺ—Ā—É –≤ –ī–Ľ–ł–Ĺ–Ĺ–ĺ–≤–ĺ–Ľ–Ĺ–ĺ–≤—É—é –ĺ–Ī–Ľ–į—Ā—ā—Ć[49].
–ė–ö-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—ā–ĺ–ī—č –į–Ĺ–į–Ľ–ł–∑–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–ė–ö-—Ā–Ņ–Ķ–ļ—ā—Ä—č –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –ł–ľ–Ķ—é—ā –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ķ –≤ —ā–į–Ī–Ľ–ł—Ü–Ķ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –Ņ–ĺ–Ľ–ĺ—Ā—č, –≤—č–∑–≤–į–Ĺ–Ĺ—č–Ķ –≤–į–Ľ–Ķ–Ĺ—ā–Ĺ—č–ľ–ł –ļ–ĺ–Ľ–Ķ–Ī–į–Ĺ–ł—Ź–ľ–ł —Ā–≤—Ź–∑–ł –°=–° –ł C-H[50]:
| –Ę–ł–Ņ—č –ļ–ĺ–Ľ–Ķ–Ī–į–Ĺ–ł–Ļ –ł –≥—Ä—É–Ņ–Ņ
|
–Ē–ł–į–Ņ–į–∑–ĺ–Ĺ, —Ā–ľ‚ąí1
|
–ü—Ä–ł–ľ–Ķ—á–į–Ĺ–ł–Ķ
|
–í–į–Ľ–Ķ–Ĺ—ā–Ĺ—č–Ķ –ļ–ĺ–Ľ–Ķ–Ī–į–Ĺ–ł—Ź —Ā–≤—Ź–∑–Ķ–Ļ C‚ąíH
|
| R2C=CH2
|
3095-3075
|
–ú–ĺ–≥—É—ā –Ĺ–į–Ī–Ľ—é–ī–į—ā—Ć—Ā—Ź –ľ—É–Ľ—Ć—ā–ł–Ņ–Ľ–Ķ—ā—č
|
| R2C=CHR
|
3045-3010
|
–Ē–ł—Ą—Ą–Ķ—Ä–Ķ–Ĺ—Ü–ł–į—Ü–ł—Ź —Ü–ł—Ā- –ł —ā—Ä–į–Ĺ—Ā- –ł–∑–ĺ–ľ–Ķ—Ä–ĺ–≤ –Ĺ–Ķ–≤–ĺ–∑–ľ–ĺ–∂–Ĺ–į
|
–Ē–Ķ—Ą–ĺ—Ä–ľ–į—Ü–ł–ĺ–Ĺ–Ĺ—č–Ķ –ļ–ĺ–Ľ–Ķ–Ī–į–Ĺ–ł—Ź —Ā–≤—Ź–∑–Ķ–Ļ C‚ąíH
|
| RCH=CH2
|
990, 910
|
|
| R,RC=CH2
|
–ĺ–ļ–ĺ–Ľ–ĺ 890
|
|
| R,RC=CHR
|
840-790
|
|
| —ā—Ä–į–Ĺ—Ā‚ÄĒRCH=CHR
|
–ĺ–ļ–ĺ–Ľ–ĺ 950
|
|
| —Ü–ł—Ā‚ąíRCH=CHR
|
730-665
|
|
–í–į–Ľ–Ķ–Ĺ—ā–Ĺ—č–Ķ –ļ–ĺ–Ľ–Ķ–Ī–į–Ĺ–ł—Ź —Ā–≤—Ź–∑–Ķ–Ļ C=–°
|
| —ā—Ä–į–Ĺ—Ā‚ąíRCH=CHR
|
–ĺ–ļ–ĺ–Ľ–ĺ 1675
|
–ü–ĺ–Ľ–ĺ—Ā—č —É–ľ–Ķ—Ä–Ķ–Ĺ–Ĺ–ĺ–Ļ –ł –≤—č—Ā–ĺ–ļ–ĺ–Ļ –ł–Ĺ—ā–Ķ–Ĺ—Ā–ł–≤–Ĺ–ĺ—Ā—ā–ł, –Ņ—Ä–ł–≥–ĺ–ī–Ĺ—č–Ķ –ī–Ľ—Ź –ł–ī–Ķ–Ĺ—ā–ł—Ą–ł–ļ–į—Ü–ł–ł –į—Ü–ł–ļ–Ľ–ł—á–Ķ—Ā–ļ–ł—Ö –ł –Ĺ–Ķ–Ĺ–į–Ņ—Ä—Ź–∂—Ď–Ĺ–Ĺ—č—Ö —Ā–ł—Ā—ā–Ķ–ľ
|
| —Ü–ł—Ā‚ąíRCH=CHR
|
–ĺ–ļ–ĺ–Ľ–ĺ 1660
|
| RCH=CR1R2
|
–ĺ–ļ–ĺ–Ľ–ĺ 1670
|
| R2C=CH2
|
–ĺ–ļ–ĺ–Ľ–ĺ 1650
|
| RCH=CH2
|
–ĺ–ļ–ĺ–Ľ–ĺ 1640
|
| C=C‚ąíC=C
|
1645-1600
|
–ü–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ –Ņ–ĺ–Ľ–ĺ—Ā—č, –Ī–ĺ–Ľ–Ķ–Ķ –ł–Ĺ—ā–Ķ–Ĺ—Ā–ł–≤–Ĺ–ĺ–Ļ —á–Ķ–ľ —É –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤, –∑–į–≤–ł—Ā–ł—ā –ĺ—ā –≥–Ķ–ĺ–ľ–Ķ—ā—Ä–ł–ł —Ā–ĺ–Ņ—Ä—Ź–∂—Ď–Ĺ–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č
|
| C=C‚ąíC=O
|
1660-1580
|
| C=C‚ąí(C=C)n
|
1650-1580
|
–ü–ĺ–Ľ–ĺ—Ā—č –ł–ľ–Ķ—é—ā –ľ—É–Ľ—Ć—ā–ł–Ņ–Ľ–Ķ—ā–Ĺ—É—é —Ā—ā—Ä—É–ļ—ā—É—Ä—É, –į –Ņ—Ä–ł –Ī–ĺ–Ľ—Ć—ą–ł—Ö n —Ā–Ľ–ł–≤–į—é—ā—Ā—Ź –≤ –ĺ–ī–Ĺ—É —ą–ł—Ä–ĺ–ļ—É—é –Ņ–ĺ–Ľ–ĺ—Ā—É
|
| ArC=C
|
–ĺ–ļ–ĺ–Ľ–ĺ 1630
|
–ü–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ –Ņ–ĺ–Ľ–ĺ—Ā—č –∑–į–≤–ł—Ā–ł—ā –ĺ—ā –Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł—Ź –ł –Ņ—Ä–ł—Ä–ĺ–ī—č –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–Ķ–Ļ
|
–Į–ú–†-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—ā–ĺ–ī—č –į–Ĺ–į–Ľ–ł–∑–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–Į–ú–†-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—á–Ķ—Ā–ļ–ł–Ķ –ľ–Ķ—ā–ĺ–ī—č –į–Ĺ–į–Ľ–ł–∑–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź—é—ā –ł–ī–Ķ–Ĺ—ā–ł—Ą–ł—Ü–ł—Ä–ĺ–≤–į—ā—Ć —Ā–ł–≥–Ĺ–į–Ľ—č –į—ā–ĺ–ľ–ĺ–≤ –≤–ĺ–ī–ĺ—Ä–ĺ–ī–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤, —ā–Ķ–ľ —Ā–į–ľ—č–ľ –Ņ–ĺ–Ľ—É—á–ł–≤ –≤–į–∂–Ĺ—É—é –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł—é –ĺ —Ā—ā—Ä—É–ļ—ā—É—Ä–Ķ —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī–ĺ–≤. –≠—ā–ł —Ā–ł–≥–Ĺ–į–Ľ—č –Ľ–Ķ–∂–į—ā –≤ –ī–ł–į–Ņ–į–∑–ĺ–Ĺ–Ķ 4-8 –ľ.–ī. –°—É—Č–Ķ—Ā—ā–≤—É–Ķ—ā —ć–ľ–Ņ–ł—Ä–ł—á–Ķ—Ā–ļ–į—Ź –∑–į–≤–ł—Ā–ł–ľ–ĺ—Ā—ā—Ć, –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź—é—Č–į—Ź –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ —ā–ĺ—á–Ĺ–ĺ –≤—č—á–ł—Ā–Ľ–ł—ā—Ć —Ā–ī–≤–ł–≥–ł –Ņ—Ä–ĺ—ā–ĺ–Ĺ–ĺ–≤ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤[51]:
őīC=C-H = 5,25 + Z–≥–Ķ–ľ + Z—Ü–ł—Ā + Z—ā—Ä–į–Ĺ—Ā
–≥–ī–Ķ Z-–į–ī–ī–ł—ā–ł–≤–Ĺ—č–Ķ –Ņ–į—Ä–į–ľ–Ķ—ā—Ä—č —ć–ļ—Ä–į–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤—É—é—Č–ł—Ö –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–Ķ–Ļ.
–ó–Ĺ–į—á–Ķ–Ĺ–ł—Ź Z –ī–Ľ—Ź –ĺ—ā–ī–Ķ–Ľ—Ć–Ĺ—č—Ö –∑–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ–Ķ–Ļ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ—č –≤ —ā–į–Ī–Ľ–ł—Ü–Ķ[51]:
| –ó–į–ľ–Ķ—Ā—ā–ł—ā–Ķ–Ľ—Ć
|
Z–≥–Ķ–ľ
|
Z—Ü–ł—Ā
|
Z—ā—Ä–į–Ĺ—Ā
|
| H
|
0,00
|
0,00
|
0,00
|
| –ź–Ľ–ļ–ł–Ľ
|
0,45
|
-0,22
|
-0,28
|
| –ź–Ľ–ļ–ł–Ľ (—Ü–ł–ļ–Ľ.)*
|
0,69
|
-0,25
|
-0,28
|
| CH2Ar
|
1,05
|
-0,29
|
-0,32
|
| CH2X (X:F, Cl, Br)
|
0,70
|
0,11
|
-0,04
|
| CH2OH
|
0,64
|
-0,01
|
-0,02
|
| CH2NH2
|
0,58
|
-0,10
|
-0,08
|
| C=C (–ł–∑–ĺ–Ľ–ł—Ä.)
|
1,00
|
-0,09
|
-0,23
|
| C=C (—Ā–ĺ–Ņ—Ä—Ź–∂.)
|
1,24
|
0,02
|
-0,05
|
| Ar
|
1,38
|
0,36
|
-0,07
|
| Cl
|
1,08
|
0,18
|
0,13
|
| Br
|
1,07
|
0,45
|
0,55
|
| OR
|
1,22
|
-1,07
|
-1,21
|
| OC(O)R
|
2,11
|
-0,35
|
-0,64
|
| CHO
|
1,02
|
0,95
|
1,17
|
| COOH
|
0,97
|
1,41
|
0,71
|
| COOR
|
0,80
|
1,18
|
0,55
|
* ‚ÄĒ –Ē–≤–ĺ–Ļ–Ĺ–į—Ź —Ā–≤—Ź–∑—Ć –ł –į–Ľ–ļ–ł–Ľ –≤—Ö–ĺ–ī—Ź—ā –≤ —Ü–ł–ļ–Ľ
–ü—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–ź–Ľ–ļ–Ķ–Ĺ—č —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –≤–į–∂–Ĺ–Ķ–Ļ—ą–ł–ľ —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł–ľ —Ā—č—Ä—Ć—Ď–ľ.
–ü—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ–Ķ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ —ć—ā–ł–Ľ–Ķ–Ĺ–į
–≠—ā–ł–Ľ–Ķ–Ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ—ā—Ā—Ź –ī–Ľ—Ź –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į —Ü–Ķ–Ľ–ĺ–≥–ĺ —Ä—Ź–ī–į —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ: –≤–ł–Ĺ–ł–Ľ—Ö–Ľ–ĺ—Ä–ł–ī–į, —Ā—ā–ł—Ä–ĺ–Ľ–į, —ć—ā–ł–Ľ–Ķ–Ĺ–≥–Ľ–ł–ļ–ĺ–Ľ—Ź, —ć—ā–ł–Ľ–Ķ–Ĺ–ĺ–ļ—Ā–ł–ī–į, —ć—ā–į–Ĺ–ĺ–Ľ–į–ľ–ł–Ĺ–ĺ–≤, —ć—ā–į–Ĺ–ĺ–Ľ–į, –ī–ł–ĺ–ļ—Ā–į–Ĺ–į, –ī–ł—Ö–Ľ–ĺ—Ä—ć—ā–į–Ĺ–į, —É–ļ—Ā—É—Ā–Ĺ–ĺ–≥–ĺ –į–Ľ—Ć–ī–Ķ–≥–ł–ī–į –ł —É–ļ—Ā—É—Ā–Ĺ–ĺ–Ļ –ļ–ł—Ā–Ľ–ĺ—ā—č[35]. –ü–ĺ–Ľ–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł–Ķ–Ļ —ć—ā–ł–Ľ–Ķ–Ĺ–į –ł –Ķ–≥–ĺ –Ņ—Ä—Ź–ľ—č—Ö –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č—Ö –Ņ–ĺ–Ľ—É—á–į—é—ā –Ņ–ĺ–Ľ–ł—ć—ā–ł–Ľ–Ķ–Ĺ, –Ņ–ĺ–Ľ–ł–≤–ł–Ĺ–ł–Ľ–į—Ü–Ķ—ā–į—ā, –Ņ–ĺ–Ľ–ł–≤–ł–Ĺ–ł–Ľ—Ö–Ľ–ĺ—Ä–ł–ī, –ļ–į—É—á—É–ļ–ł –ł —Ā–ľ–į–∑–ĺ—á–Ĺ—č–Ķ –ľ–į—Ā–Ľ–į.
–ú–ł—Ä–ĺ–≤–ĺ–Ķ –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–ĺ —ć—ā–ł–Ľ–Ķ–Ĺ–į —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ķ—ā –Ņ–ĺ—Ä—Ź–ī–ļ–į 100 –ľ–Ľ–Ĺ —ā–ĺ–Ĺ–Ĺ –≤ –≥–ĺ–ī[52] (–Ņ–ĺ –ī–į–Ĺ–Ĺ—č–ľ –Ĺ–į 2005 –≥–ĺ–ī: 107 –ľ–Ľ–Ĺ —ā–ĺ–Ĺ–Ĺ[53]).
–ü—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ–Ķ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ –Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ–į
–ü—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ –≤ –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–Ķ—ā—Ā—Ź, –≤ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–ľ, –ī–Ľ—Ź —Ā–ł–Ĺ—ā–Ķ–∑–į –Ņ–ĺ–Ľ–ł–Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ–į (62 % –Ņ—Ä–ĺ—Ü–Ķ–Ĺ—ā–į –≤—Ā–Ķ–≥–ĺ –≤—č–Ņ—É—Ā–ļ–į–Ķ–ľ–ĺ–≥–ĺ –ĺ–Ī—ä—Ď–ľ–į[54]). –Ę–į–ļ–∂–Ķ –ł–∑ –Ĺ–Ķ–≥–ĺ –Ņ–ĺ–Ľ—É—á–į—é—ā –ļ—É–ľ–ĺ–Ľ, –ĺ–ļ–ł—Ā—Ć –Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ–į, –į–ļ—Ä–ł–Ľ–ĺ–Ĺ–ł—ā—Ä–ł–Ľ, –ł–∑–ĺ–Ņ—Ä–ĺ–Ņ–į–Ĺ–ĺ–Ľ, –≥–Ľ–ł—Ü–Ķ—Ä–ł–Ĺ, –ľ–į—Ā–Ľ—Ź–Ĺ—č–Ļ –į–Ľ—Ć–ī–Ķ–≥–ł–ī[35].
–í –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –ľ–ł—Ä–ĺ–≤—č–Ķ –ľ–ĺ—Č–Ĺ–ĺ—Ā—ā–ł –Ņ–ĺ –≤—č–Ņ—É—Ā–ļ—É –Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ–į —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź—é—ā –ĺ–ļ–ĺ–Ľ–ĺ 70 –ľ–Ľ–Ĺ —ā–ĺ–Ĺ–Ĺ –≤ –≥–ĺ–ī[54]. –ü–ĺ –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑–į–ľ —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–≤, –Ņ–ĺ—ā—Ä–Ķ–Ī–Ĺ–ĺ—Ā—ā—Ć –≤ –Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ–Ķ –≤ –Ī–Ľ–ł–∂–į–Ļ—ą–Ķ–ľ –Ī—É–ī—É—Č–Ķ–ľ –Ī—É–ī–Ķ—ā —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –Ņ—Ä–Ķ–≤—č—ą–į—ā—Ć –ĺ–Ī—ä—Ď–ľ—č –Ķ–≥–ĺ –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į, –Ņ—Ä–ł—á—Ď–ľ, –ĺ–∂–ł–ī–į–Ķ—ā—Ā—Ź, —á—ā–ĺ –ļ 2010 –≥–ĺ–ī—É –ĺ–Ī—ä—Ď–ľ –Ķ–≥–ĺ –ľ–ł—Ä–ĺ–≤–ĺ–≥–ĺ –≤—č–Ņ—É—Ā–ļ–į –ī–ĺ—Ā—ā–ł–≥–Ĺ–Ķ—ā 90 –ľ–Ľ–Ĺ —ā–ĺ–Ĺ–Ĺ[55].
–ü—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ–Ķ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ –Ņ—Ä–ĺ—á–ł—Ö –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
–Ď—É—ā–ł–Ľ–Ķ–Ĺ—č –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—é—ā –ī–Ľ—Ź –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–į –Ī—É—ā–į–ī–ł–Ķ–Ĺ–į, –ł–∑–ĺ–Ņ—Ä–Ķ–Ĺ–į, –Ņ–ĺ–Ľ–ł–ł–∑–ĺ–Ī—É—ā–ł–Ľ–Ķ–Ĺ–į, –Ī—É—ā–ł–Ľ–ļ–į—É—á—É–ļ–į, –ľ–Ķ—ā–ł–Ľ—ć—ā–ł–Ľ–ļ–Ķ—ā–ĺ–Ĺ–į –ł –Ņ—Ä[56].
–ė–∑–ĺ–Ī—É—ā–ł–Ľ–Ķ–Ĺ ‚ÄĒ —Ā—č—Ä—Ć—Ď –ī–Ľ—Ź –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –Ī—É—ā–ł–Ľ–ļ–į—É—á—É–ļ–į, –ł–∑–ĺ–Ņ—Ä–Ķ–Ĺ–į, —ā—Ä–Ķ—ā-–Ī—É—ā–į–Ĺ–ĺ–Ľ–į; –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ—ā—Ā—Ź –ī–Ľ—Ź –į–Ľ–ļ–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ą–Ķ–Ĺ–ĺ–Ľ–ĺ–≤ –Ņ—Ä–ł —Ā–ł–Ĺ—ā–Ķ–∑–Ķ –ü–ź–í. –ē–≥–ĺ —Ā–ĺ–Ņ–ĺ–Ľ–ł–ľ–Ķ—Ä—č —Ā –Ī—É—ā–Ķ–Ĺ–į–ľ–ł –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—é—ā –ļ–į–ļ –Ņ—Ä–ł—Ā–į–ī–ļ–ł –ļ –ľ–į—Ā–Ľ–į–ľ –ł –≥–Ķ—Ä–ľ–Ķ—ā–ł–ļ–ł.
–í—č—Ā—ą–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ—č –°10‚ąí–°18 –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—é—ā –Ņ—Ä–ł —Ā–ł–Ĺ—ā–Ķ–∑–Ķ –ü–ź–í, –į —ā–į–ļ–∂–Ķ –ī–Ľ—Ź –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł—Ź –≤—č—Ā—ą–ł—Ö —Ā–Ņ–ł—Ä—ā–ĺ–≤.
–°–ľ. —ā–į–ļ–∂–Ķ
–Ē–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –≤–Ĺ–Ķ—ą–Ĺ–ł–Ķ –ł—Ā—ā–ĺ—á–Ĺ–ł–ļ–ł
–ě–Ī—Č–ł–Ķ –Ľ–Ķ–ļ—Ü–ł–ł –Ņ–ĺ —Ö–ł–ľ–ł–ł –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
- –ė–Ľ–Ľ—é—Ā—ā—Ä–į—ā–ł–≤–Ĺ—č–Ķ –ľ–į—ā–Ķ—Ä–ł–į–Ľ—č –Ľ–Ķ–ļ—Ü–ł–Ļ –Ņ–ĺ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł –Ņ—Ä–ĺ—Ą–Ķ—Ā—Ā–ĺ—Ä–į –Ě–Ķ–Ĺ–į–Ļ–ī–Ķ–Ĺ–ļ–ĺ –í. –ď., –Ľ–Ķ–ļ—Ü–ł—Ź ‚ĄĖ 7 (–ź–Ľ–ļ–Ķ–Ĺ—č. –°—ā—Ä–ĺ–Ķ–Ĺ–ł–Ķ, –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ł–Ķ, —Ä–Ķ–į–ļ—Ü–ł–ĺ–Ĺ–Ĺ–į—Ź —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā—Ć.)
- –ė–Ľ–Ľ—é—Ā—ā—Ä–į—ā–ł–≤–Ĺ—č–Ķ –ľ–į—ā–Ķ—Ä–ł–į–Ľ—č –Ľ–Ķ–ļ—Ü–ł–Ļ –Ņ–ĺ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł –Ņ—Ä–ĺ—Ą–Ķ—Ā—Ā–ĺ—Ä–į –Ě–Ķ–Ĺ–į–Ļ–ī–Ķ–Ĺ–ļ–ĺ –í. –ď., –Ľ–Ķ–ļ—Ü–ł—Ź ‚ĄĖ 8 (–ź–Ľ–ļ–Ķ–Ĺ—č. –†–Ķ–į–ļ—Ü–ł–ĺ–Ĺ–Ĺ–į—Ź —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā—Ć.)
- –ė–Ľ–Ľ—é—Ā—ā—Ä–į—ā–ł–≤–Ĺ—č–Ķ –ľ–į—ā–Ķ—Ä–ł–į–Ľ—č –Ľ–Ķ–ļ—Ü–ł–Ļ –Ņ–ĺ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł –Ņ—Ä–ĺ—Ą–Ķ—Ā—Ā–ĺ—Ä–į –Ě–Ķ–Ĺ–į–Ļ–ī–Ķ–Ĺ–ļ–ĺ –í. –ď., –Ľ–Ķ–ļ—Ü–ł—Ź ‚ĄĖ 9 (–ź–Ľ–ļ–Ķ–Ĺ—č. –†–Ķ–į–ļ—Ü–ł–ĺ–Ĺ–Ĺ–į—Ź —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā—Ć.)
- –ö—É—Ä—Ü –ź. –õ., –õ–ł–≤–į–Ĺ—Ü–ĺ–≤ –ú. –í., –õ–ł–≤–į–Ĺ—Ü–ĺ–≤–į –õ. –ė. –ź–Ľ–ļ–Ķ–Ĺ—č (–ß–į—Ā—ā—Ć I). –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–į–ļ—É–Ľ—Ć—ā–Ķ—ā –ú–ď–£, 1998 –≥–ĺ–ī.
- –ö—É—Ä—Ü –ź. –õ., –õ–ł–≤–į–Ĺ—Ü–ĺ–≤ –ú. –í., –õ–ł–≤–į–Ĺ—Ü–ĺ–≤–į –õ. –ė. –ź–Ľ–ļ–Ķ–Ĺ—č (–ß–į—Ā—ā—Ć II). –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–į–ļ—É–Ľ—Ć—ā–Ķ—ā –ú–ď–£, 1999 –≥–ĺ–ī.
–£—á–Ķ–Ī–Ĺ–į—Ź –Ľ–ł—ā–Ķ—Ä–į—ā—É—Ä–į
- –Ě–Ķ–Ļ–Ľ–į–Ĺ–ī –ě. –Į. –ď–Ľ–į–≤–į II. –ź–Ľ–ļ–Ķ–Ĺ—č // –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź: –£—á–Ķ–Ī. –ī–Ľ—Ź —Ö–ł–ľ. –≤—É–∑–ĺ–≤. ‚ÄĒ –ú.: ¬ę–í—č—Ā—ą–į—Ź —ą–ļ–ĺ–Ľ–į¬Ľ, 1990. ‚ÄĒ –°. 102‚ÄĒ130. ‚ÄĒ ISBN 5-06-001471-1.
- –†–ĺ–Ī–Ķ—Ä—ā—Ā –Ē–∂., –ö–į—Ā–Ķ—Ä–ł–ĺ –ú. –ď–Ľ–į–≤–į 6. –ź–Ľ–ļ–Ķ–Ĺ—č. –°—ā—Ä—É–ļ—ā—É—Ä–į, —Ā–Ņ–Ķ–ļ—ā—Ä—č –ł —Ā—ā–Ķ—Ä–Ķ–ĺ–ł–∑–ĺ–ľ–Ķ—Ä–ł—Ź. –ď–Ľ–į–≤–į 7. –ź–Ľ–ļ–Ķ–Ĺ—č. –†–Ķ–į–ļ—Ü–ł–ł –ī–≤–ĺ–Ļ–Ĺ—č—Ö —É–≥–Ľ–Ķ—Ä–ĺ–ī-—É–≥–Ľ–Ķ—Ä–ĺ–ī–Ĺ—č—Ö —Ā–≤—Ź–∑–Ķ–Ļ // –ě—Ā–Ĺ–ĺ–≤—č –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł / –ü–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –į–ļ–į–ī–Ķ–ľ–ł–ļ–į –Ě–Ķ—Ā–ľ–Ķ—Ź–Ĺ–ĺ–≤–į –ź.–Ě.. ‚ÄĒ 2-–Ķ, –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ–Ĺ–ĺ–Ķ. ‚ÄĒ –ú.: –ú–ł—Ä, 1978. ‚ÄĒ –Ę. 1. ‚ÄĒ –°. 171‚ÄĒ235.
- –†–Ķ—É—ā–ĺ–≤ –ě. –ź., –ö—É—Ä—Ü –ź. –õ., –Ď—É—ā–ł–Ĺ –ö. –ü. –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź. –í 4 —á–į—Ā—ā—Ź—Ö. ‚ÄĒ 3-–Ķ –ł–∑–ī–į–Ĺ–ł–Ķ. ‚ÄĒ –ú.: –Ď–ł–Ĺ–ĺ–ľ. –õ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–ł—Ź –∑–Ĺ–į–Ĺ–ł–Ļ, 2007. ‚ÄĒ –Ę. 1. ‚ÄĒ 568 —Ā. ‚ÄĒ ISBN 978-5-94774-613-6.
- –Ę—Ä–į–≤–Ķ–Ĺ—Ć –í. –§. –ď–Ľ–į–≤–į 5. –ź–Ľ–ļ–Ķ–Ĺ—č // –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź: –£—á–Ķ–Ī–Ĺ–ł–ļ –ī–Ľ—Ź –≤—É–∑–ĺ–≤: –í 2 —ā / –í. –§. –Ę—Ä–į–≤–Ķ–Ĺ—Ć. ‚ÄĒ –ú.: –ė–ö–¶ ¬ę–ź–ļ–į–ī–Ķ–ľ–ļ–Ĺ–ł–≥–į¬Ľ, 2004. ‚ÄĒ –Ę. 1. ‚ÄĒ –°. 237‚ÄĒ305. ‚ÄĒ ISBN 5-94628-171-2.
–ú–Ķ—Ö–į–Ĺ–ł–∑–ľ—č —Ä–Ķ–į–ļ—Ü–ł–Ļ —Ā —É—á–į—Ā—ā–ł–Ķ–ľ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤
- –ú–į—Ä—á –Ē–∂. –ď–Ľ–į–≤–į 15. –†–Ķ–į–ļ—Ü–ł–ł –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ļ –ļ—Ä–į—ā–Ĺ—č–ľ —Ā–≤—Ź–∑—Ź–ľ —É–≥–Ľ–Ķ—Ä–ĺ–ī-—É–≥–Ľ–Ķ—Ä–ĺ–ī. –ď–Ľ–į–≤–į 16. –†–Ķ–į–ļ—Ü–ł–ł –Ņ—Ä–ł—Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź –ļ –ļ—Ä–į—ā–Ĺ—č–ľ —Ā–≤—Ź–∑—Ź–ľ —É–≥–Ľ–Ķ—Ä–ĺ–ī-–≥–Ķ—ā–Ķ—Ä–ĺ–į—ā–ĺ–ľ. // –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź. –†–Ķ–į–ļ—Ü–ł–ł, –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ł —Ā—ā—Ä—É–ļ—ā—É—Ä–į. –£–≥–Ľ—É–Ī–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –ļ—É—Ä—Ā –ī–Ľ—Ź —É–Ĺ–ł–≤–Ķ—Ä—Ā–ł—ā–Ķ—ā–ĺ–≤ –ł —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –≤—É–∑–ĺ–≤: –≤ 4-—Ö —ā–ĺ–ľ–į—Ö / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ., –Ņ–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –ė. –ü. –Ď–Ķ–Ľ–Ķ—Ü–ļ–ĺ–Ļ. ‚ÄĒ –ú.: –ú–ł—Ä, 1988. ‚ÄĒ –Ę. 3. ‚ÄĒ –°. 132‚ÄĒ430.
- –°–į–Ļ–ļ—Ā –ü. –ú–Ķ—Ö–į–Ĺ–ł–∑–ľ—č —Ä–Ķ–į–ļ—Ü–ł–Ļ –≤ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ.,–Ņ–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –í. –§. –Ę—Ä–į–≤–Ķ–Ĺ—Ź. ‚ÄĒ 4-–Ķ –ł–∑–ī. ‚ÄĒ –ú.: –•–ł–ľ–ł—Ź, 1991. ‚ÄĒ 448 —Ā. ‚ÄĒ ISBN 5-7245-0191-0.
–ė—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ –≤ –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł
- –≠—ā–ł–Ľ–Ķ–Ĺ–ĺ–≤–ĺ–Ķ –Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī—Ā—ā–≤–ĺ –≤ –°–Ě–ď: —Ä–Ķ–į–ļ—ā–ĺ—Ä—č –ł –ļ–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä—č
- –ü–ĺ–Ľ–ł–ĺ–Ľ–Ķ—Ą–ł–Ĺ—č: –Ĺ–ĺ–≤—č–Ķ —ā–Ķ—Ö–Ĺ–ĺ–Ľ–ĺ–≥–ł–ł –ł —Ä—č–Ĺ–ĺ–ļ
–ü—Ä–ł–ľ–Ķ—á–į–Ĺ–ł—Ź
- ‚ÜĎ Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names 2013. Chapter P-15.1.7.2.2 (–į–Ĺ–≥–Ľ.) / Henri A. Favre, Warren H. Powell, International Union of Pure and Applied Chemistry. ‚ÄĒ Cambridge: Royal Soc. of Chemistry [u.a.], 2014. ‚ÄĒ 1568 p. ‚ÄĒ ISBN 978-0-85404-182-4. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 14 —Ą–Ķ–≤—Ä–į–Ľ—Ź 2024 –≥–ĺ–ī–į.
- ‚ÜĎ 1 2 3 4 5
–Ę—Ä–į–≤–Ķ–Ĺ—Ć –í.–§. –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź: –£—á–Ķ–Ī–Ĺ–ł–ļ –ī–Ľ—Ź –≤—É–∑–ĺ–≤: –í 2 —ā / –í.–§.–Ę—Ä–į–≤–Ķ–Ĺ—Ć. ‚ÄĒ –ú.: –ė–ö–¶ ¬ę–ź–ļ–į–ī–Ķ–ľ–ļ–Ĺ–ł–≥–į¬Ľ, 2004. ‚ÄĒ –Ę. 1. ‚ÄĒ 727 —Ā. ‚ÄĒ ISBN 5-94628-171-2.
- ‚ÜĎ 1 2 3 –ú–į–∑–į–Ľ–ĺ–≤ –õ.–Ě. –≠–Ľ–Ķ–ļ—ā—Ä–ĺ–Ĺ–Ĺ–ĺ-—Ā—ā—Ä—É–ļ—ā—É—Ä–Ĺ—č–Ķ —Ą–į–ļ—ā–ĺ—Ä—č –≤ —ć–ļ—Ā—ā—Ä–į–ļ—Ü–ł–ł (–Ĺ–Ķ–ĺ–Ņ—Ä.). –Ė—É—Ä–Ĺ–į–Ľ —Ā—ā—Ä—É–ļ—ā—É—Ä–Ĺ–ĺ–Ļ —Ö–ł–ľ–ł–ł. –ė–Ě–• –°–ě –†–ź–Ě (17 –ĺ–ļ—ā—Ź–Ī—Ä—Ź 2002). –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 24 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ –ł–∑ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ–į 20 —Ā–Ķ–Ĺ—ā—Ź–Ī—Ä—Ź 2008 –≥–ĺ–ī–į.
- ‚ÜĎ –°–Ľ—É—á–į–Ļ–Ĺ—č–Ķ –ĺ—ā–ļ—Ä—č—ā–ł—Ź. –≠—ā–ł–Ľ–Ķ–Ĺ (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ó–į–Ĺ–ł–ľ–į—ā–Ķ–Ľ—Ć–Ĺ–į—Ź —Ö–ł–ľ–ł—Ź. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 19 –ľ–į—Ź 2007 –≥–ĺ–ī–į.
- ‚ÜĎ –ě—ā–ļ—Ä—č—ā–ł–Ķ —ć—ā–ł–Ľ–Ķ–Ĺ–į (–Ĺ–Ķ–ĺ–Ņ—Ä.) (pdf). –ě—ā–ļ—Ä—č—ā–ł—Ź –≤ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł –ł –Ī–ł–ĺ—Ö–ł–ľ–ł–ł. –ē–ī–ł–Ĺ–į—Ź –ļ–ĺ–Ľ–Ľ–Ķ–ļ—Ü–ł—Ź —Ü–ł—Ą—Ä–ĺ–≤—č—Ö –ĺ–Ī—Ä–į–∑–ĺ–≤–į—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ä–Ķ—Ā—É—Ä—Ā–ĺ–≤. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 2 –ł—é–Ĺ—Ź 2013 –≥–ĺ–ī–į.
- ‚ÜĎ 1 2
–ú–Ķ–Ĺ—ą—É—ā–ļ–ł–Ĺ –Ě. –ě—á–Ķ—Ä–ļ—ä —Ä–į–∑–≤–ł—ā—Ė—Ź —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö—ä –≤–ĺ–∑–∑—Ä“Ć–Ĺ—Ė–Ļ. ‚ÄĒ –°-–ü–Ķ—ā–Ķ—Ä–Ī—É—Ä–≥—ä: –Ę–ł–Ņ. –í.–Ē–Ķ–ľ–į–ļ–ĺ–≤–į, 1888. ‚ÄĒ –°. 252‚ÄĒ264.
- ‚ÜĎ
–§–ł–≥—É—Ä–ĺ–≤—Ā–ļ–ł–Ļ –Ě.–ź. –ė—Ā—ā–ĺ—Ä–ł—Ź —Ö–ł–ľ–ł–ł: –£—á–Ķ–Ī. –Ņ–ĺ—Ā–ĺ–Ī–ł–Ķ –ī–Ľ—Ź —Ā—ā—É–ī–Ķ–Ĺ—ā–ĺ–≤ –Ņ–Ķ–ī. –ł–Ĺ-—ā–ĺ–≤ –Ņ–ĺ —Ö–ł–ľ. –ł –Ī–ł–ĺ–Ľ. —Ā–Ņ–Ķ—Ü. ‚ÄĒ –ú.: –ü—Ä–ĺ—Ā–≤–Ķ—Č–Ķ–Ĺ–ł–Ķ, 1979. ‚ÄĒ –°. 102.
- ‚ÜĎ
–°–ĺ–Ľ–ĺ–≤—Ć–Ķ–≤ –ģ. –ė. –ė—Ā—ā–ĺ—Ä–ł—Ź —Ö–ł–ľ–ł–ł: –†–į–∑–≤–ł—ā–ł–Ķ —Ö–ł–ľ–ł–ł —Ā –ī—Ä–Ķ–≤–Ĺ–Ķ–Ļ—ą–ł—Ö –≤—Ä–Ķ–ľ–Ķ–Ĺ –ī–ĺ –ļ–ĺ–Ĺ—Ü–į XIX –≤. –ü–ĺ—Ā–ĺ–Ī–ł–Ķ –ī–Ľ—Ź —É—á–ł—ā–Ķ–Ľ–Ķ–Ļ. ‚ÄĒ 2-–Ķ –ł–∑–ī., –Ņ–Ķ—Ä–Ķ—Ä–į–Ī. ‚ÄĒ –ú.: –ü—Ä–ĺ—Ā–≤–Ķ—Č–Ķ–Ĺ–ł–Ķ, 1983. ‚ÄĒ –°. 208.
- ‚ÜĎ –í –Ņ—Ä–ł—Ä–ĺ–ī–Ķ —Ā—É—Č–Ķ—Ā—ā–≤—É–Ķ—ā –Ī–ĺ–Ľ—Ć—ą–ĺ–Ķ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ —Ā –ī–≤–ĺ–Ļ–Ĺ—č–ľ–ł —Ā–≤—Ź–∑—Ź–ľ–ł, –Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä —ā–Ķ—Ä–Ņ–Ķ–Ĺ—č –ł–Ľ–ł –ļ–į—Ä–ĺ—ā–ł–Ĺ–ĺ–ł–ī—č, –ĺ–ī–Ĺ–į–ļ–ĺ –ł—Ö –ĺ—ā–Ĺ–ĺ—Ā—Ź—ā –ļ –ĺ—ā–ī–Ķ–Ľ—Ć–Ĺ—č–ľ –ļ–Ľ–į—Ā—Ā–į–ľ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ –ł –≤ –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ļ —Ā—ā–į—ā—Ć–Ķ –ĺ–Ĺ–ł –Ĺ–Ķ —Ä–į—Ā—Ā–ľ–į—ā—Ä–ł–≤–į—é—ā—Ā—Ź.
- ‚ÜĎ 1 2 –í—Ä–Ķ–ī–Ĺ—č–Ķ –≤–Ķ—Č–Ķ—Ā—ā–≤–į. –Ě–Ķ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ć–Ĺ—č–Ķ —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī—č —ć—ā–ł–Ľ–Ķ–Ĺ–ĺ–≤–ĺ–≥–ĺ —Ä—Ź–ī–į (–į–Ľ–ļ–Ķ–Ĺ—č) (–Ĺ–Ķ–ĺ–Ņ—Ä.). –Ě–ĺ–≤—č–Ļ —Ā–Ņ—Ä–į–≤–ĺ—á–Ĺ–ł–ļ —Ö–ł–ľ–ł–ļ–į –ł —ā–Ķ—Ö–Ĺ–ĺ–Ľ–ĺ–≥–į. Chemanalytica.com. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 16 —Ź–Ĺ–≤–į—Ä—Ź 2012 –≥–ĺ–ī–į.
- ‚ÜĎ –Ě–Ķ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ć–Ĺ—č–Ķ, –ł–Ľ–ł –Ĺ–Ķ–Ĺ–į—Ā—č—Č–Ķ–Ĺ–Ĺ—č–Ķ, —É–≥–Ľ–Ķ–≤–ĺ–ī–ĺ—Ä–ĺ–ī—č —Ä—Ź–ī–į —ć—ā–ł–Ľ–Ķ–Ĺ–į (–į–Ľ–ļ–Ķ–Ĺ—č) (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź. Chemistry.narod.ru. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 28 –ľ–į—Ź 2013 –≥–ĺ–ī–į.
- ‚ÜĎ –°–≤–ĺ–Ļ—Ā—ā–≤–į –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ. –°–Ņ—Ä–į–≤–ĺ—á–Ĺ–ł–ļ. / –ü–ĺ–ī. —Ä–Ķ–ī. –ź. –ź. –ü–ĺ—ā–Ķ—Ö–ł–Ĺ–į. –õ. –•–ł–ľ–ł—Ź. ‚ÄĒ 1984. ‚ÄĒ 520 —Ā.
- ‚ÜĎ The Nobel Prize in Chemistry 1979 (–į–Ĺ–≥–Ľ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 25 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 22 –į–≤–≥—É—Ā—ā–į 2011 –≥–ĺ–ī–į.
- ‚ÜĎ 1 2 3 4 5 6
–ú–į—Ä—á –Ē–∂. –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź. –†–Ķ–į–ļ—Ü–ł–ł, –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ł —Ā—ā—Ä—É–ļ—ā—É—Ä–į. –£–≥–Ľ—É–Ī–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –ļ—É—Ä—Ā –ī–Ľ—Ź —É–Ĺ–ł–≤–Ķ—Ä—Ā–ł—ā–Ķ—ā–ĺ–≤ –ł —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –≤—É–∑–ĺ–≤: –≤ 4-—Ö —ā–ĺ–ľ–į—Ö = Advanced organic chemistry. Reactions, Mechanisms and Structure / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ., –Ņ–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –ė.–ü.–Ď–Ķ–Ľ–Ķ—Ü–ļ–ĺ–Ļ. ‚ÄĒ –ú.: –ú–ł—Ä, 1988. ‚ÄĒ –Ę. 3. ‚ÄĒ 459 —Ā.
- ‚ÜĎ –†–ĺ–Ī–Ķ—Ä—ā—Ā –Ē–∂., –ö–į—Ā–Ķ—Ä–ł–ĺ –ú. –ě—Ā–Ĺ–ĺ–≤—č –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł = Basic principles of organic chemistry / –ü–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –į–ļ–į–ī–Ķ–ľ–ł–ļ–į –Ě–Ķ—Ā–ľ–Ķ—Ź–Ĺ–ĺ–≤–į –ź.–Ě.. ‚ÄĒ 2-–Ķ, –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ–Ĺ–ĺ–Ķ. ‚ÄĒ –ú.: –ú–ł—Ä, 1978. ‚ÄĒ –Ę. 1. ‚ÄĒ –°. 227‚ÄĒ228.
- ‚ÜĎ –ö–ĺ–Ĺ–ī–į–ļ–ĺ–≤–į —Ä–Ķ–į–ļ—Ü–ł—Ź // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 2. ‚ÄĒ –°. 887‚ÄĒ888.
- ‚ÜĎ –ú–į—Ä—á –Ē–∂. –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź. –†–Ķ–į–ļ—Ü–ł–ł, –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ł —Ā—ā—Ä—É–ļ—ā—É—Ä–į. –£–≥–Ľ—É–Ī–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –ļ—É—Ä—Ā –ī–Ľ—Ź —É–Ĺ–ł–≤–Ķ—Ä—Ā–ł—ā–Ķ—ā–ĺ–≤ –ł —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –≤—É–∑–ĺ–≤: –≤ 4-—Ö —ā–ĺ–ľ–į—Ö = Advanced organic chemistry. Reactions, Mechanisms and Structure / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ., –Ņ–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –ė.–ü.–Ď–Ķ–Ľ–Ķ—Ü–ļ–ĺ–Ļ. ‚ÄĒ –ú.: –ú–ł—Ä, 1988. ‚ÄĒ –Ę. 2. ‚ÄĒ 504 —Ā.
- ‚ÜĎ –ö—É—Ä—Ü –ź –õ., –õ–ł–≤–į–Ĺ—Ü–ĺ–≤ –ú.–í., –õ–ł–≤–į–Ĺ—Ü–ĺ–≤–į –õ.–ė. –ö–į—Ä–Ī–Ķ–Ĺ—č –ł –ļ–į—Ä–Ī–Ķ–Ĺ–ĺ–ł–ī—č (—Ä–į–∑–ī–Ķ–Ľ 4.7.) (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ź–Ľ–ļ–Ķ–Ĺ—č (—á–į—Ā—ā—Ć II). –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–į–ļ—É–Ľ—Ć—ā–Ķ—ā –ú–ď–£. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 5 –ī–Ķ–ļ–į–Ī—Ä—Ź 2011 –≥–ĺ–ī–į.
- ‚ÜĎ –ú–į—Ä—á –Ē–∂. –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź. –†–Ķ–į–ļ—Ü–ł–ł, –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ł —Ā—ā—Ä—É–ļ—ā—É—Ä–į. –£–≥–Ľ—É–Ī–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –ļ—É—Ä—Ā –ī–Ľ—Ź —É–Ĺ–ł–≤–Ķ—Ä—Ā–ł—ā–Ķ—ā–ĺ–≤ –ł —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –≤—É–∑–ĺ–≤: –≤ 4-—Ö —ā–ĺ–ľ–į—Ö = Advanced organic chemistry. Reactions, Mechanisms and Structure / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ., –Ņ–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –ė.–ü.–Ď–Ķ–Ľ–Ķ—Ü–ļ–ĺ–Ļ. ‚ÄĒ –ú.: –ú–ł—Ä, 1988. ‚ÄĒ –Ę. 1. ‚ÄĒ –°. 253.
- ‚ÜĎ 1 2 3 4 –õ–ł –Ē–∂. –ė–ľ–Ķ–Ĺ–Ĺ—č–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł. –ú–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ä–Ķ–į–ļ—Ü–ł–Ļ = Name reactions / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ. –í.–ú.–Ē–Ķ–ľ—Ć—Ź–Ĺ–ĺ–≤–ł—á. ‚ÄĒ –ú.: –Ď–ė–Ě–ě–ú. –õ–į–Ī–ĺ—Ä–į—ā–ĺ—Ä–ł—Ź –∑–Ĺ–į–Ĺ–ł–Ļ, 2006. ‚ÄĒ 456 —Ā. ‚ÄĒ ISBN 5-94774-368-X.
- ‚ÜĎ 1 2 3 –ö—É—Ä—Ü –ź –õ., –õ–ł–≤–į–Ĺ—Ü–ĺ–≤ –ú.–í., –õ–ł–≤–į–Ĺ—Ü–ĺ–≤–į –õ.–ė. –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ā–≤–ĺ–Ļ—Ā—ā–≤–į –į–Ľ–ļ–Ķ–Ĺ–ĺ–≤ (—Ä–į–∑–ī–Ķ–Ľ 4.) (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ź–Ľ–ļ–Ķ–Ĺ—č (—á–į—Ā—ā—Ć II). –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–į–ļ—É–Ľ—Ć—ā–Ķ—ā –ú–ď–£. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 20 —Ź–Ĺ–≤–į—Ä—Ź 2012 –≥–ĺ–ī–į.
- ‚ÜĎ –ú–į–ļ–ļ–≤–ł–Ľ–Ľ–ł–Ĺ –§. –Ē–∂. –ď–ĺ–ľ–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–Ķ –≥–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –≤ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł = Homogeneous hydrogenation in organic chemistry / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ. –Ě.–ú.–õ–ĺ–Ļ–ľ–į. ‚ÄĒ –ú.: –•–ł–ľ–ł—Ź, 1980. ‚ÄĒ 160 —Ā.
- ‚ÜĎ –í–ĺ–Ľ—Ź-–¶–ł–≥–Ľ–Ķ—Ä–į —Ä–Ķ–į–ļ—Ü–ł—Ź // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 1. ‚ÄĒ –°. 824‚ÄĒ825.
- ‚ÜĎ 1 2 3 4 5 –•–Ķ–Ļ–Ĺ—Ā –ź. –ú–Ķ—ā–ĺ–ī—č –ĺ–ļ–ł—Ā–Ľ–Ķ–Ĺ–ł—Ź –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ: –ź–Ľ–ļ–į–Ĺ—č, –į–Ľ–ļ–Ķ–Ĺ—č, –į–Ľ–ļ–ł–Ĺ—č –ł –į—Ä–Ķ–Ĺ—č = Methods for the oxidation of organic compounds: Alkanes, Alkenes, Alkynes and Arenes / –ü–Ķ—Ä–Ķ–≤–ĺ–ī —Ā –į–Ĺ–≥–Ľ., –Ņ–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –ė.–ü. –Ď–Ķ–Ľ–Ķ—Ü–ļ–ĺ–Ļ. ‚ÄĒ –ú.: –ú–ł—Ä, 1988. ‚ÄĒ 400 —Ā. ‚ÄĒ ISBN 5-03-000149-2.
- ‚ÜĎ –ü—Ä–ł–Ľ–Ķ–∂–į–Ķ–≤–į —Ä–Ķ–į–ļ—Ü–ł—Ź // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 4. ‚ÄĒ –°. 169.
- ‚ÜĎ 1 2 3
–§–į–Ľ—Ć–Ī–Ķ –ģ. –°–ł–Ĺ—ā–Ķ–∑ –Ĺ–į –ĺ—Ā–Ĺ–ĺ–≤–Ķ –ĺ–ļ–ł—Ā–ł —É–≥–Ľ–Ķ—Ä–ĺ–ī–į / –ü–Ķ—Ä. —Ā –Ĺ–Ķ–ľ. ‚ÄĒ –õ., 1971.
- ‚ÜĎ The Nobel Prize in Chemistry 2005 (–į–Ĺ–≥–Ľ.). Nobel Prize in Chemistry. The Official Web Site of the Nobel Foundation. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 22 –į–≤–≥—É—Ā—ā–į 2011 –≥–ĺ–ī–į.
- ‚ÜĎ –ú–Ķ—Ö–į–Ĺ–ł–∑–ľ —Ä–Ķ–į–ļ—Ü–ł–ł –ľ–Ķ—ā–į—ā–Ķ–∑–ł—Ā–į (–Ĺ–Ķ–ĺ–Ņ—Ä.) (jpg). –°–į–Ļ—ā –∂—É—Ä–Ĺ–į–Ľ–į "–Ě–į—É–ļ–į –ł –∂–ł–∑–Ĺ—Ć". –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. (–Ĺ–Ķ–ī–ĺ—Ā—ā—É–Ņ–Ĺ–į—Ź —Ā—Ā—č–Ľ–ļ–į)
- ‚ÜĎ –Ě–ĺ–Ī–Ķ–Ľ–Ķ–≤—Ā–ļ—É—é –Ņ—Ä–Ķ–ľ–ł—é –Ņ–ĺ —Ö–ł–ľ–ł–ł –Ņ—Ä–ł—Ā—É–ī–ł–Ľ–ł –ė–≤—É –®–į–≤–Ķ–Ĺ—É, –†–ĺ–Ī–Ķ—Ä—ā—É –ď—Ä—É–Ī–Ī—Ā—É –ł –†–ł—á–į—Ä–ī—É –®—Ä–ĺ–ļ—É (–Ĺ–Ķ–ĺ–Ņ—Ä.). –Ě–ĺ–≤–ĺ—Ā—ā–ł. Lenta.ru (5 –ĺ–ļ—ā—Ź–Ī—Ä—Ź 2005). –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 27 —Ā–Ķ–Ĺ—ā—Ź–Ī—Ä—Ź 2011 –≥–ĺ–ī–į.
- ‚ÜĎ –ú–Ķ—ā–į—ā–Ķ–∑–ł—Ā –≤ –≤–ĺ–ī–Ĺ–ĺ–Ļ —Ā—Ä–Ķ–ī–Ķ (–Ĺ–Ķ–ĺ–Ņ—Ä.). –Ě–ĺ–≤–ĺ—Ā—ā–ł —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ĺ–į—É–ļ–ł. –ü–ĺ—Ä—ā–į–Ľ Chemport.ru (9 —Ą–Ķ–≤—Ä–į–Ľ—Ź 2008). –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 25 –ľ–į—Ä—ā–į 2013 –≥–ĺ–ī–į.
- ‚ÜĎ
–ě–Ľ–Ķ—Ą–ł–Ĺ—č // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 3. ‚ÄĒ –°. 737‚ÄĒ740.
- ‚ÜĎ –Ē–Ķ–≥–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –į–Ľ–ļ–į–Ĺ–ĺ–≤ (—Ä–į–∑–ī–Ķ–Ľ 2.5.3.) (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ė–Ĺ—ā–Ķ—Ä–į–ļ—ā–ł–≤–Ĺ—č–Ļ –ľ—É–Ľ—Ć—ā–ł–ľ–Ķ–ī–ł–į —É—á–Ķ–Ī–Ĺ–ł–ļ "–ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź". –°–į–ľ–į—Ä—Ā–ļ–ł–Ļ –ď–£, –ö–į—Ą–Ķ–ī—Ä–į –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ, –Ī–ł–ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ł –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ –ł–∑ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ–į 28 –ĺ–ļ—ā—Ź–Ī—Ä—Ź 2011 –≥–ĺ–ī–į.
- ‚ÜĎ –ź–Ľ–ļ–Ķ–Ĺ—č –ł –į–Ľ–ļ–į–ī–ł–Ķ–Ĺ—č –ł–∑ –į–Ľ–ļ–į–Ĺ–ĺ–≤ (–Ĺ–Ķ–ĺ–Ņ—Ä.). –Ě–Ķ—Ą—ā–Ķ—Ö–ł–ľ–ł—Ź. Chemistry.narod.ru. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ –ł–∑ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ–į 16 –ĺ–ļ—ā—Ź–Ī—Ä—Ź 2009 –≥–ĺ–ī–į.
- ‚ÜĎ –ö–į—ā–į–Ľ–ł–∑–į—ā–ĺ—Ä—č –ī–Ķ–≥–ł–ī—Ä–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 2. ‚ÄĒ –°. 670‚ÄĒ671.
- ‚ÜĎ 1 2 3 4 5
–Ě–Ķ–Ļ–Ľ–į–Ĺ–ī –ě. –Į. –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź: –£—á–Ķ–Ī. –ī–Ľ—Ź —Ö–ł–ľ. –≤—É–∑–ĺ–≤. ‚ÄĒ –ú.: ¬ę–í—č—Ā—ą–į—Ź —ą–ļ–ĺ–Ľ–į¬Ľ, 1990. ‚ÄĒ 750 —Ā. ‚ÄĒ ISBN 5-06-001471-1.
- ‚ÜĎ
–ú–į—ā—Ć—Ď –Ė., –ü–į–Ĺ–ł–ļ–ĺ –†., –í–Ķ–Ļ–Ľ—Ć-–†–Ķ–Ļ–Ĺ–į–Ľ—Ć –Ė. –ė–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –ł –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ —Ą—É–Ĺ–ļ—Ü–ł–Ļ –≤ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–ľ —Ā–ł–Ĺ—ā–Ķ–∑–Ķ = L'amenagement fonctionnel en synthese organique / –ü–Ķ—Ä–Ķ–≤–ĺ–ī —Ā —Ą—Ä–į–Ĺ—Ü—É–∑—Ā–ļ–ĺ–≥–ĺ –°.–°. –ģ—Ą–ł—ā–į. ‚ÄĒ –ú.: ¬ę–ú–ł—ĬĽ, 1980. ‚ÄĒ –°. 169.
- ‚ÜĎ
–ö–Ķ—Ä—Ä–ł –§, –°–į–Ĺ–ī–Ī–Ķ—Ä–≥ –†. –ö–Ĺ–ł–≥–į –Ņ–Ķ—Ä–≤–į—Ź. –°—ā—Ä—É–ļ—ā—É—Ä–į –ł –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č // –£–≥–Ľ—É–Ī–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –ļ—É—Ä—Ā –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ., –Ņ–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –Ņ—Ä–ĺ—Ą. –í.–ú.–ü–ĺ—ā–į–Ņ–ĺ–≤–į. ‚ÄĒ –ú.: –•–ł–ľ–ł—Ź, 1981. ‚ÄĒ –°. 54‚ÄĒ59.
- ‚ÜĎ –•–ĺ—Ä–Ĺ–Ķ—Ä–į —Ä–Ķ–į–ļ—Ü–ł—Ź // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 5. ‚ÄĒ –°. 606‚ÄĒ607.
- ‚ÜĎ –†–Ķ–į–ļ—Ü–ł—Ź –ß—É–≥–į–Ķ–≤–į (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ė–ľ–Ķ–Ĺ–Ĺ—č–Ķ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł. –ė—Ä–ļ—É—ā—Ā–ļ–ł–Ļ –≥–ĺ—Ā—É–ī–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ļ —É–Ĺ–ł–≤–Ķ—Ä—Ā–ł—ā–Ķ—ā. –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–į–ļ—É–Ľ—Ć—ā–Ķ—ā. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ –ł–∑ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ–į 19 –į–Ņ—Ä–Ķ–Ľ—Ź 2011 –≥–ĺ–ī–į.
- ‚ÜĎ –ź–ľ–ľ–ĺ–Ĺ–ł–Ķ–≤—č–Ķ —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł—Ź // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 1. ‚ÄĒ –°. 278‚ÄĒ280.
- ‚ÜĎ 1 2 3 –ú–į—Ä—á –Ē–∂. –ě—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–į—Ź —Ö–ł–ľ–ł—Ź. –†–Ķ–į–ļ—Ü–ł–ł, –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ł —Ā—ā—Ä—É–ļ—ā—É—Ä–į. –£–≥–Ľ—É–Ī–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –ļ—É—Ä—Ā –ī–Ľ—Ź —É–Ĺ–ł–≤–Ķ—Ä—Ā–ł—ā–Ķ—ā–ĺ–≤ –ł —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł—Ö –≤—É–∑–ĺ–≤: –≤ 4-—Ö —ā–ĺ–ľ–į—Ö = Advanced organic chemistry. Reactions, Mechanisms and Structure / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ., –Ņ–ĺ–ī —Ä–Ķ–ī–į–ļ—Ü–ł–Ķ–Ļ –ė.–ü.–Ď–Ķ–Ľ–Ķ—Ü–ļ–ĺ–Ļ. ‚ÄĒ –ú.: –ú–ł—Ä, 1988. ‚ÄĒ –Ę. 4. ‚ÄĒ –°. 49‚ÄĒ53.
- ‚ÜĎ –†–Ķ–į–ļ—Ü–ł—Ź –Ď—É—Ä–ī–į (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ė–ľ–Ķ–Ĺ–Ĺ—č–Ķ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ä–Ķ–į–ļ—Ü–ł–ł. –ė—Ä–ļ—É—ā—Ā–ļ–ł–Ļ –≥–ĺ—Ā—É–ī–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ļ —É–Ĺ–ł–≤–Ķ—Ä—Ā–ł—ā–Ķ—ā. –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–į–ļ—É–Ľ—Ć—ā–Ķ—ā. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ –ł–∑ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ–į 10 –į–Ņ—Ä–Ķ–Ľ—Ź 2013 –≥–ĺ–ī–į.
- ‚ÜĎ –Ď—ć–ľ—Ą–ĺ—Ä–ī–į-–°—ā–ł–≤–Ķ–Ĺ—Ā–į —Ä–Ķ–į–ļ—Ü–ł—Ź // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 1. ‚ÄĒ –°. 658.
- ‚ÜĎ –Ē—Ź–ī—á–Ķ–Ĺ–ļ–ĺ –í.–ü., –ź–Ĺ–ī—Ä–Ķ—Ā—é–ļ –ź.–Ě, –Ď–Ķ–Ľ–ĺ–≥–Ľ–į–∑–ļ–ł–Ĺ–į –ē.–ö., –Ď—Ä—É—Ā–ĺ–≤–į –ď.–ü. –ė—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ –∑–į—Č–ł—ā–Ĺ—č—Ö –≥—Ä—É–Ņ–Ņ –≤ —Ā–ł–Ĺ—ā–Ķ–∑–Ķ (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ü–Ľ–į–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ľ–Ĺ–ĺ–≥–ĺ—Ā—ā–į–ī–ł–Ļ–Ĺ—č—Ö —Ā–ł–Ĺ—ā–Ķ–∑–ĺ–≤. –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–į–ļ—É–Ľ—Ć—ā–Ķ—ā –ú–ď–£ (2003). –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 25 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 4 –ľ–į—Ä—ā–į 2016 –≥–ĺ–ī–į.
- ‚ÜĎ –ü–Ķ—Ä–ļ–ł–Ĺ–į —Ä–Ķ–į–ļ—Ü–ł—Ź // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 3. ‚ÄĒ –°. 965‚ÄĒ966.
- ‚ÜĎ –®—Ä–į–Ļ–Ĺ–Ķ—Ä –†., –§—Ć—é–∑–ĺ–Ĺ –†., –ö—Ď—Ä—ā–ł–Ĺ –Ē., –ú–ĺ—Ä—Ä–ł–Ľ–Ľ –Ę. –ė–ī–Ķ–Ĺ—ā–ł—Ą–ł–ļ–į—Ü–ł—Ź –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ (–Ĺ–Ķ–ĺ–Ņ—Ä.). –•–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ļ –ļ–į—ā–į–Ľ–ĺ–≥. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 25 –Ĺ–ĺ—Ź–Ī—Ä—Ź 2019 –≥–ĺ–ī–į.
- ‚ÜĎ –í—É–Ľ—Ć—Ą—Ā–ĺ–Ĺ –Ě.–°., –ó–į–ł–ļ–ł–Ĺ –í.–ď., –ú–ł–ļ–į—Ź –ź.–ė. –ú–į—Ā—Ā-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ–ľ–Ķ—ā—Ä–ł—Ź –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–ĺ–Ķ–ī–ł–Ĺ–Ķ–Ĺ–ł–Ļ. ‚ÄĒ –•–ł–ľ–ł—Ź. ‚ÄĒ –ú., 1986. ‚ÄĒ –°. 31.
- ‚ÜĎ
–ú–ł–ļ–į—Ź –ź.–ė., –°–ľ–Ķ—ā–į–Ĺ–ł–Ĺ –í.–ė., –ó–į–ł–ļ–ł–Ĺ –í.–ď. // –°–Ķ—Ä–ł—Ź —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź : –°–Ī. ‚ÄĒ –ė–∑–≤–Ķ—Ā—ā–ł—Ź –ź–Ě –°–°–°–†, 1982. ‚ÄĒ –°. 2214.
- ‚ÜĎ
–ö–į–∑–ł—Ü—č–Ĺ–į –õ.–ź., –ö—É–Ņ–Ľ–Ķ—ā—Ā–ļ–į—Ź –Ě.–Ď. –ü—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –£–§-, –ė–ö- –ł –Į–ú–†-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł–ł –≤ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł. ‚ÄĒ –ú.: –í—č—Ā—ą–į—Ź —ą–ļ–ĺ–Ľ–į, 1971. ‚ÄĒ –°. 66‚ÄĒ67.
- ‚ÜĎ
–Ď—Ä–į—É–Ĺ –Ē., –§–Ľ–ĺ–Ļ–ī –ź., –°–Ķ–Ļ–Ĺ–∑–Ī–Ķ—Ä–ł –ú. –°–Ņ–Ķ–ļ—ā—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—Ź –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –≤–Ķ—Č–Ķ—Ā—ā–≤ = Organic Spectroscopy / –ü–Ķ—Ä. —Ā –į–Ĺ–≥–Ľ. –ź. –ź. –ö–ł—Ä—é—ą–ļ–ł–Ĺ–į. ‚ÄĒ –ú.: –ú–ł—Ä, 1992. ‚ÄĒ –°. 50. ‚ÄĒ ISBN 5-03-002111-6.
- ‚ÜĎ 1 2
–ė–ĺ–Ĺ–ł–Ĺ –Ď.–ė., –ē—Ä—ą–ĺ–≤ –Ď.–ź., –ö–ĺ–Ľ—Ć—Ü–ĺ–≤ –ź.–ė. –Į–ú–†-—Ā–Ņ–Ķ–ļ—ā—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—Ź –≤ –ĺ—Ä–≥–į–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ł / –ü–ĺ–ī —Ä–Ķ–ī. –ē—Ä—ą–ĺ–≤–į –Ď.–ź.. ‚ÄĒ 2-–Ķ –ł–∑–ī., –Ņ–Ķ—Ä–Ķ—Ä–į–Ī. ‚ÄĒ –õ.: –•–ł–ľ–ł—Ź, 1983. ‚ÄĒ –°. 157‚ÄĒ158.
- ‚ÜĎ –ü—Ä–ĺ–≥–Ĺ–ĺ–∑ —Ä—č–Ĺ–ļ–į —ć—ā–ł–Ľ–Ķ–Ĺ–į (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ö–ĺ–Ĺ—ä—é–Ĺ–ļ—ā—É—Ä–į. –Ę–ĺ–≤–į—Ä—č –ł —Ä—č–Ĺ–ļ–ł. –†–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ł–Ļ –¶–Ķ–Ĺ—ā—Ä –≤–Ĺ–Ķ—ą–Ĺ–Ķ–Ļ —ā–ĺ—Ä–≥–ĺ–≤–Ľ–ł. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 30 –ī–Ķ–ļ–į–Ī—Ä—Ź 2012 –≥–ĺ–ī–į.
- ‚ÜĎ –≠—ā–ł–Ľ–Ķ–Ĺ, —ć—ā–Ķ–Ĺ (–Ĺ–Ķ–ĺ–Ņ—Ä.). –°—ā–į—ā—Ć–ł –ĺ –≥–į–∑–į—Ö. –ö–ĺ–ľ–Ņ–į–Ĺ–ł—Ź "–Ě–ė–ė –ö–ú". –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 8 –ī–Ķ–ļ–į–Ī—Ä—Ź 2011 –≥–ĺ–ī–į.
- ‚ÜĎ 1 2 –ú–ł—Ä–ĺ–≤–ĺ–Ļ —Ä—č–Ĺ–ĺ–ļ –Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ–į (–Ĺ–Ķ–ĺ–Ņ—Ä.). Ssa.ru. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 20 —Ā–Ķ–Ĺ—ā—Ź–Ī—Ä—Ź 2011 –≥–ĺ–ī–į.
- ‚ÜĎ –ú–Ķ—ā–į—ā–Ķ–∑–ł—Ā –ĺ–Ľ–Ķ—Ą–ł–Ĺ–ĺ–≤: —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ—č–Ļ –Ņ—É—ā—Ć –ļ –Ņ–ĺ–Ľ–ł–Ņ—Ä–ĺ–Ņ–ł–Ľ–Ķ–Ĺ—É (–Ĺ–Ķ–ĺ–Ņ—Ä.). –ź–Ĺ–į–Ľ–ł—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –Ņ–ĺ—Ä—ā–į–Ľ —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–ĺ–ľ—č—ą–Ľ–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł: –Ě–ĺ–≤—č–Ķ —Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł–Ķ —ā–Ķ—Ö–Ĺ–ĺ–Ľ–ĺ–≥–ł–ł. –Ē–į—ā–į –ĺ–Ī—Ä–į—Č–Ķ–Ĺ–ł—Ź: 22 –ł—é–Ľ—Ź 2009. –ź—Ä—Ö–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–ĺ 15 –ł—é–Ľ—Ź 2014 –≥–ĺ–ī–į.
- ‚ÜĎ –Ď—É—ā–Ķ–Ĺ—č // –•–ł–ľ–ł—á–Ķ—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź / –ď–Ľ–į–≤–Ĺ—č–Ļ —Ä–Ķ–ī–į–ļ—ā–ĺ—Ä –ė. –õ. –ö–Ĺ—É–Ĺ—Ź–Ĺ—Ü. ‚ÄĒ –ú.: ¬ę–°–ĺ–≤–Ķ—ā—Ā–ļ–į—Ź —ć–Ĺ—Ü–ł–ļ–Ľ–ĺ–Ņ–Ķ–ī–ł—Ź¬Ľ, 1988. ‚ÄĒ –Ę. 1. ‚ÄĒ –°. 638‚ÄĒ640.





-9-tricosene.svg)






















{kind=link}