![]() 维基百科
维基百科中的醫學内容
仅供参考,並
不能視作專業意見。如需獲取醫療幫助或意見,请咨询专业人士。詳見
醫學聲明。
希佩尔-林道综合征(Von Hippel–Lindau disease,VHL综合征)是一种罕见的常染色体显性遗传性疾病[1],表现为血管母细胞瘤累及小脑、脊髓、肾脏以及视网膜。其若干病变包括肾脏血管瘤、肾细胞癌以及嗜铬细胞瘤等。疾病是因位于染色体3P25.3的VHL抑癌基因发生突变所致。[2]
临床表现和症状
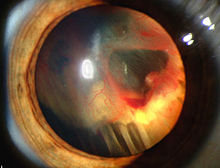 裂隙灯照片显示VHL疾病的视网膜分离
裂隙灯照片显示VHL疾病的视网膜分离
VHL的临床表现和症状包括血管瘤、血管母细胞瘤、嗜铬细胞瘤、肾细胞癌,胰腺囊肿(胰腺浆液性囊腺瘤)以及咖啡牛奶斑[3]。37.2%的VHL患者表现为血管瘤,且通常累及视网膜,由此产生的失明十分常见。其他脏器也会受累,由此带来的中风、心脏病、心血管疾病也相当多见。[2]
遗传学
希佩尔-林道综合征是因位于3号染色体短臂(3P25-26)的VHL抑癌基因突变所致。
 VHL综合征是常染色体显性遗传模式
VHL综合征是常染色体显性遗传模式
只是一个等位基因上的VHL基因突变,那么细胞内仍能产生有功能的VHL蛋白,肿瘤也不会发生。因为疾病的产生需要两个等位基因同时突变,所以第二个等位基因必需在体内至少一个细胞内发生突变,这就是所谓的二次突变假说。如果第二个等位基因发生突变,细胞就失去了可以产生功能性VHL蛋白的基因。VHL蛋白的丢失会导致有肿瘤特性的希佩尔- 林道综合征的发生。
遗传性VHL基因突变占到了80%。而大约20%的病例,是由于基因在生殖细胞(卵子或精子)形成时或在胎儿生长早期发生新的突变所致,这种情况非常罕见,因为在一个细胞中,原先均正常的两个等位基因同时发生突变的概率很低。无论是新的突变还是遗传性突变,前面提及的第二次突变对于肿瘤的发生是不可缺的。
疾病的发病年龄、对器官的影响和病变程度在不同患者中差别很大。这提示了第二次突变可发生在不同类型的细胞中,且发生在人生的不同阶段。
治疗
如今没有方法可以逆转患者VHL基因的突变。尽管如此,早期识别VHL综合征特征性的临床表现并进行治疗,这能从本质上减少并发症的发生并提高患者的生活质量。
历史
 对于VHL疾病最早的描述
对于VHL疾病最早的描述
德国眼科医生Eugen von Hippel在1904年第一个描述了眼球内血管瘤的病例。[4]Arvid Lindau在1927年描述了小脑及脊髓内血管瘤的病例。[5] 疾病也因此用两人的姓氏命名。
人物
一些McCoy家族(即美国阿巴拉契亚和Hatfield家族有世仇的McCoy家族)以及Elliott家族的后裔患有VHL。在一篇美联社的报道中,一位来自范德堡大学的内分泌学家推测,Hatfield家族和McCoy家族间潜在的敌意,有一部分可能是由于VHL疾病导致的。报道指出,McCoy家族有脾气暴躁的倾向,因为家族中的许多人患有嗜铬细胞瘤,产生过多的肾上腺素,从而导致暴躁的脾气。[6] 但是,嗜铬细胞瘤产生大量的肾上腺素,更多表现为惊恐发作而不是暴力袭击。未经治疗的患者会有罹患心血管疾病,心脏病以及中风的危险。只有大约20%的VHL患者有嗜铬细胞瘤。[7]
命名
希佩尔-林道综合征还有其他不常见的命名,包括:视网膜血管瘤,囊肿性视网膜血管瘤(小脑及视网膜内血管瘤样囊肿形成),家族性小脑-视网膜血管瘤,小脑视网膜血管母细胞瘤,视网膜小脑血管瘤。
参考文献
外部链接
参见
|
|---|
| 1-10 | |
|---|
| 11-20 | |
|---|
| 21-30 | |
|---|
| 31-40 | |
|---|
| 41-50 | |
|---|
| 51-60 | |
|---|
| 61-70 | |
|---|
| 71-80 | |
|---|
| 81-86 | |
|---|
|



